(1978)
advertisement
")
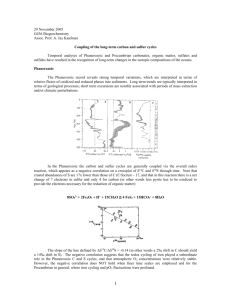
-- ^---LI1L~F-- -^.x^l^- II*II~ II^I~ -)~11.1-^_11 1-L. _LI ANALYSES OF THE SULFUR SYSTEM IN WATERS FROM THE GALAPAGOS RIDGE HYDROTHERMAL VENTS by SARAH S. HUESTED S.B., Massachusetts Institute of Technology (1978) SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER, 1979 Signature of Author De , of/Tarth & Planetary Sciences Certified by ..... Thesis Supervisor Accepted by .............. .................................... arary ChairmaYhr I MIT Tr I4US INSTTUIE I IRRARIES Sciences Committee ~-~--~LL~II -- --. 1IIYI ll111 .(r-._r ~-i.-~L~--_l~l) I -2ANALYSES OF THE SULFUR SYSTEM IN WATERS FROM THE GALAPAGOS RIDGE HYDROTHERMAL VENTS by SARAH S. HUESTED Submitted to the Department of Earth and Planetary Sciences on September 1979 in partial fulfillment of the requirements for the Degree of Master of Science ABSTRACT Samples collected from the Galapagos hydrothermal vents show a decrease in sulfate with temperature resulting in a flux of 5.3 x 1012 mol/year from the ocean assuming that these vents are representative of a global process. Sulfide concentrations increased with temperature (flux of 2.6 x 10 11-2.6 x 1012 mol/year to the ocean). Reduced species of sulfur including sulfite, thiosulfate, trithionate and tetrathionate were found in minimal amounts, if at all. A substantial portion of the iodine reactive species (otherwise unaccounted for) was hypothesized to be elemental sulfur. Reduced sulfur species were not found in the overlying water column. Name of Supervisor: Professor John M. Edmond, M.I.T. .. - - 0 -3ACKNOWLEDGEMENTS I am grateful to John M. Edmond for being my advisor and providing me with insight and suggestions for my research. Special thanks are due the many people who helped me accomplish the research work, including Bob Stallard, Barry Grant, Russ McDuff, and Chris Measures. Thanks are also due Loretta Tocio for typing a major part of this thesis. I was supported by funds from ONR and NSF. ... r ,.~..r-lll- -- --x~ -;-^-r---~x-I ll ..._-~---rul-r~--l-~-L-LI~-LIICT -Y-~L-~-- _~ -4TABLE OF CONTENTS Page Number Title Page......................... ..... Abstract... ........................ 000. 0 2 Acknowledgements ................... .. 0.. 3 Table of Contents.................. .0.. 4 ..... 6 1 List of Figures .................... List of Tables ..................... CHAPTER 1 - Introduction........... CHAPTER 2 - Experimental Section... CHAPTER 3 - Results and Discussion. References ....................... ..... 15 ~1_. ~ ---- -I---------------c --*v---L------~l ---- '-I----------- ----*----II --- --------ur ~------------- .1-~IISS131Y+IYLI IIIII~II lrP~~IXI~ I*YU~YIWIYIIIIC liT-~e;~ Y^--~--YFC -5- List of Figures Page Number Figure 1.1 Hydrothermal Reduction and Oxida- 11 tion of Sulfur 1.2 Oxidation Reaction Pathways - 13 Acidic and Neutral Solutions 2.1 Sulfide Analytical Procedure 2.2 0-3 2.3 Sulfite Analytical Procedure 24 2.4 Thiosulfate, Tetrathionate, and 29 MM. Sulfide versus Absorbance 18 21 Trithionate Analytical Procedure 2.5 Thiocyanate versus Absorbance, 31 Method C, (CuSO4 ) 2.6 PRA Preparation 37 3.1 Sulfide versus Silica 50 3.2 Sulfide versus Oxygen 52 3.3 Sulfate versus Silica 55 3.4 Sulfite versus Silica 57 3.5 Thiosulfate versus Silica 59 3.6 Iodine Reactive Species versus Silica 61 3.7 Iodine Reactive Species Minus Sulfide, 64 Thiosulfate, and Sulfite Versus Silica .~ , .~....1... ._I~.-,i~~-~----- -3~-~~ ------- - --~~ -~--------- I--. ~e -.-,.r~l-ir~ ~ iL ~.~lr..-,~. .,~x.~-I---------- L-~rre---------- ~. ..~,~x---..~~ -6List of Tables Page Number Table 2.1 Hydrothermal Vent Locations 16 2.2 Hydrocast Locations 17 2.3 Sulfide: Reagent Concentrations 20 and Dilutions 2.4 Sulfide Precision 23 2.5 Cyanolysis Reactions Involving 27 Thiosulfate, Tetrathionate, and Trithionate 2.6 Tetrathionate and Trithionate 33 Precision 2.7 Summary of Method Standard 34 Errors 3.1 Concentrations of Sulfide, Sulfite, 42 Thiosulfate, Trithionate, Tetrathionate, Iodine Reactive Species, Oxygen, and Silica for the Galapagos Vents 3.2 Gradients and Fluxes for Sulfide and Sulfate 47 x l-~-p--lr-.-. ..irr~-- - -...~.._^........~~.~~.I~llc --- -I ..... ~.. --. -7- CHAPTER 1 INTRODUCTION i~-rXCr -I IIII^L~---~- r ----- i --- - rsu ru I,-,,--, Y- 1~ -1-11-1. - Ma L~L*~*~y~l~--~-~.r--~-- -8Over geologic time, sulfur chemistry in the oceans is controlled by reactions occurring between water, rock Under and sediment phases (Goldhaber and Kaplan, 1974). surface temperatures and pressures, for instance, it is thermodynamically favorable for sulfate to be reduced by organic matter to sulfide in the absence of oxygen. However, this reaction is not observed, unless there is biological intervention. Biologically mediated reduction of sulfate might occur in the water column in areas of the ocean where the oxygen concentration approaches zero (e.g., the eastern tropical Pacific). This phenomena has not been observed, however, probably because bacteria preferentially reduce nitrate and nitrite before sulfate (Brewer, 1975, Cline and Richards, 1972). Chemical reduction of sulfate can occur in processes such as reaction (1) when seawater and basalt react at high + -2 11 Fe 2 SiO 4 + SO 4 + 4H = 7 Fe304 + FeS2 + 11 SiO 2 + 2H20 fayalite (1). magnetite pyrite temperature, as in the case of ridge crest hydrothermal systems (Bonatti, 1975), (Figure 1.1). In experiments in which basalt and seawater were reacted at 300 0 C, much of the sulfate present was incorporated in anhydrite, CaSO 4, and some pyrite with perhaps 10% being reduced to aqueous sulfide. In many of these experiments, the total reduced 1I1 I 1 ..~-r.,~~~.,~ ~...I-..~ ~~ ..,..~...~~~.1.~.,.~4 I _II_ __....., .....~.~..~~,~.I~~~_. U.sl ------- --r~-~-i~~ -- -u~~s~~-hpyuxllllL-L-LIL^~-L1C-- -9sulfur measured exceeded the amount of total sulfur available from seawater, indicating that sulfur was also being leached from the rock (Mottl, et al., 1979). Pyrite was not found in hydrothermally altered pillow basalts from the Mid-Atlantic Ridge which led to the conclusion that the reduction of sulfate must be a minor process relative to the total water flux through the crust (Humphris, et al., 1978). Another possible source for reduced sulfur in the hydrothermal waters are volcanic gases liberated beneath spreading centers that might be dissolved in the ascending seawater (Bonatti, 1975). With these discrepancies in the literature between laboratory experiments, field observations, and theory, a more detailed look into the sulfur chemistry of hydrothermal waters is needed. Seawater, when it reacts with basalt at the ridge crest, is heated to about 300 oC, decreases in pH, the result of magnesium fixation, and potential electron acceptors (oxygen, nitrate, sulfate, etc.) are consumed resulting in a reducing solution containing hydrogen sulfide (Edmond, et al., 1979a). Ambient water entrained along faults and cracks in the crust would lead to oxygenated water mixing with these reduced fluids as they rise or to oxidation of some of the pyrite (FeS2 ). Large amounts of sulfur-oxidizing bacteria living in and around the vents would also contribute to the oxidation of the hydrothermal -C--"----Y--e~~ -~---c-~-- x.---~..,..... -III-CL .I ..l.-a----- ----l-----lsLIL--- -10solution (Corliss, et al., 1979), (Figure 1.1). With the many redox reactions possible for sulfur under the above conditions, it is possible that some of the intermediate species might be measured in the hydrothermal fluids (Figure 1.2). Thermodynamic calculations indicate that sulfate, sulfide, and/or elemental sulfur would be the dominant species depending on the pH and pE. However, the system is not at thermodynamic equilibrium, so that some of the intermediate thermodynamically unstable species might be metastable for a sufficient period of time to be sampled and measured. Many variables, such as pH, initial concentration of reduced sulfur, catalysts and inhibitors, oxygen concentration, and relative reaction Based on the rates would influence the products found. experimental work of Nelson, et al., 1977, Cline and Richards, 1969, and Chen and Morris, 1972, the intermediate species that might be expected would include elemental sulfur, thiosulfate, polythionate (in particular tetrathionate and trithionate), and,-possibly, sulfite. Therefore by measuring the concentrations of these species, as well as those of sulfide and sulfate, and, as a cross check, the species that react with iodine (H2 S, S2 0 3 , SO 3, polysulfides, and possibly So stabilized by S2 0 3 or polythionates), greater insight into the sulfur cycle in the Galapagos hydrothermal waters would be obtained. --~~~~-LI - --- ~--. -- ---- C-~----- -11- Figure 1.1 Hydrothermal Reduction and Oxidation of Sulfur I I I -..EL_^I~I~Y~-~-~-I~-~~ l-lll^-.L~----XI~-l_^.~_ Il^.~l~----- -~---i.. . -13- Figure 1.2 Oxidation Reaction Pathways - Acidic and Neutral Solutions (Nelson, et al., 1977) -Z os Os 0s -Z 0 S9.-0 1 9 0 cs I EOS k - s -z c c os -~~ 0 zS Sz z Os f zEosle q IF IIIP~--Csll~/ll(lllll~_I --- -- - -- _~---- LI^~IIIP J---_ ~-PI~ ii)- .Il~ P1- ~ll^ls)~tllllr^I~_ 1..~. -15- CHAPTER 2 EXPERIMENTAL SECTION .I-~ ~. LI-.I--II~LY -- --~--~.~._ .. ......~-~s~JI, ~.~~- .~.^I--II- ~~......,- ^XI-^- III^ ~ .~Y-Y~.-X__II-I"LLill'-_.-^II-. ~--"-"----~---I-4~-L I i^- , ~-~-, ----- -16Sampling The Galapagos hydrothermal waters were collected using the sampling system described by J.B. Corliss, et al., 1979, mounted on the deep-sea submersible, Alvin. "Rose Garden," areas were sampled; Three vent "East of Eden," and "Mussel Beds" (Table 2.1). Table 2.1 Dive # Position Vent area Lon. Lat. ' Mussel Beds 0047.5 ' N. 8609.0 Rose Garden 0048.3 ' N. 86014.3 East of Eden 0047 ' N. 8602.5 ' W. ' W. W. 898, 902, 904 899, 900, 903, 905 901 The samplers were backfilled with nitrogen as samples were removed. Samples for sulfur species analyses were collected in ground glass stoppered reagent bottles using a plastic tube held at the bottom of the container. The sample was then allowed to flow into the bottle and overflow to avoid oxygen entrainment. Hydrocast samples from approximately the same area (Table 2.2) were collected using five liter OSU bottles. . ^~ 11-1-1---~1^-~--~-.rms1"----~ 11 ~~~ ~ _~ ~_~____ ~~~~~_~-LrmqYL~^ .l.IrrIPlm-.*P VI~I--~-L-~C~_lj~^L - liis~sP -17Table 2.2 Stn # 2012.1 86 Lon. Lat. ' N. 135 0038.8' N. 139 0047.7 ' N. 8502.2 ' W. 8604.8' W. 8607.7' W. Analysis All colorimetric measurements were made using a PerkinElmer 55E spectrophotometer with a 1 cm. quartz cell, except for sulfide measurements in the 1 - 3 pM.range which were made using a 4 cm. quartz cell. Preparation of reagents is summarized on pages 28-32. Sulfide Hydrogen sulfide was determined using the spectrophotometric methylene blue method (Cline, 1969) with slight modifications. N,N-dimethyl-p-phenylenediamine-sulfate (Eastman Kodak No. 1333) and ferric chloride are reacted in 6 N. hydrochloric acid to produce the reagent. The sample is added to the reagent. The procedure is optimized for a particular concentration range by changing the reagent concentrations and sample dilution (Table 2.3). The presence of sulfide is indicated by an intense blue color, the absorbance of which is measured at 670 nm. (Figure 2.1). .~-~ 1 _r ^__ II~LL.*II -i.l.i-l.-~L --rrXII.--Y;I.~.Cr^-.I r^arn irtp-.-x-l ;I--* irrr~ l----lirr~.----.. ..1_ - ~~.~__1___1_~,~.^-~-----r.-.~.--- -18- Figure 2.1 Sulfide Analytical Procedure -illi_. ___rlll____~__L_*____3 ~- ~W-- . -L~IIPII~YII~1~ IPII*Ui- I~II~PY~ -xr--~-r;-~----~ ^--L"i~-i-i---- - -19Figure 2.1 Ssample 1 - I 3 p M. H2 S 3 - 40 VM. H 2 S 40 - I 250 lM. H 2 S add 5 ml. sample to 0.4 ml. reagent 1-3 add 5 ml. sample to 0.4 ml. reagent 3-40 add 0.4 ml. sample to 0.4 ml. reagent 40-250 wait 20 min. wait 20 min. wait 20 min. measure Abs. at 670 nm. in 4 cm. cell - ~----- - -~~ --~1' -"111----1-1-;"'Xp-~~~-"l~^~--~-- -----^-Ir-l-~-r^---~rr~li-1L1--^X-XIXL*~ --I s(-~------ IIYI li--~ _I-~1L-CI~I~-- -20Table 2.3 Sulfide conc. Diamine conc. Umoles/liter g./500 ml. Ferric conc. g./500 ml. Dil. factor ml. : ml. path length (cm.) 1 - 3 0.5 0.75 1 : 1 4 3 - 40 2.0 3.0 1 : 1 1 250 8.0 12.0 2 : 25 1 40 - For sulfide concentrations expected to border on two ranges (e.g. 40 pM. sulfide), samples were treated by both procedures. The concentration of the sample was then determined by comparing it with the standard curve covering the correct range. The standards were prepared from sodium sulfide using distilled water that had been freshly boiled, cooled, and then bubbled with nitrogen gas to remove dissolved oxygen and carbon dioxide. The sulfide crystals were washed to remove oxidation products, dried, and dissolved in the water to give an approximately 0.1 N. concentration of sulfide. This solution was then standardized iodometrically (Budd & Bewick, 1952). Samples were compared to standard curves fitted by linear regression for the concentration ranges involved. (Figure 2.2). The detection limit was 1 pM. sulfide The precision at the 95% level of confidence (Table 2.4) was constant over each of the concentration ranges used. -- Ilp-l-1 l~-^. ~iU~-L -IL-.^~l^*sryY-ttl~y----------YL rll_ -21- Figure 2.2 0-3 pM. Sulfide versus Absorbance InIIUI~-LIIX 4 h -22- 3 "e 2 IL. @0 1 1 0 -a 20 40 60 80 a -L - - 100 120 ABSORBANCE 140 1 1 160 180 1- 200 220 llll- 1IL Lli L*IYIIII~YLYYrYIPIII~~YYPI*IIPLI-_I~ i-ili~ -~~ ~-..Y_.C* ill-~_ (.^. - -- ~- I~- 1_1 LI L----l.. ~-_ILI .~1II.LIBYI -_j .~i-- ~I.^_II-*-1-I-~(IF-l~ -23Table 2.4 Sulfide conc. Umol-/liter Precision mol/liter 1 - 3 + 0.05 3 - 40 + 0.9 + 29 40 - 250 Sulfate Sulfate measurements were performed by Russ McDuff using the polarographic method of G.W. Luther and A.L. Meyerson (1975). The standard error was + 0.2% at the 95% confidence level (A2). Iodine Reactive Species The iodine reactive species present were determined iodometrically by adding an excess of approximately 0.1 N. iodine and back titrating the excess iodine with 0.102 N. thiosulfate (Vogel, 1961). thiosulfate daily. The iodine was standardized with A seawater blank (approximately 46 equiv/liter of iodine) was subtracted from the samples. standard error was approximately + 65 95% confidence level. The eq/l of 12 at the This large error could have been reduced by using less concentrated solutions of iodine and thiosulfate for the titration. Sulfite The colorimetric method of Scaringelli, et al., 1967, with modifications was used for sulfite determinations (Fig 2.3). --l_~nui~-~--c_---_---rr----~i-Y~-i--~-^.I~ i--_~_-_--_-----r I ~)~LIII^~LIII-lypm_-o~xr-_~r~-ri -laa^-- ------r~L_.^ ..ll._L-.----.. ... -24- Figure 2.3 Sulfite Analytical Procedure ~ ~~P~-a~~.~d~- -- -I---r-~r,- . I~ 1 -l add -25Figure 2.3 add to 1 ml. HgC1 2 wait 10 min. wait 30 min. Y-ill~X--IY-~~ ~ ~ pCII11.11I-~--.-.-.-I.-l- ..-_--.---- -26Sulfite reacts with formaldehyde and pararosaniline to form an intense violet color in acid solution. A 0.2 M. mercuric chloride solution (stabilizes sulfite from air oxidation by formation of dichlorosulfitomercurate) was used instead of 0.04 M. HgCl 4 because of the high chloride concentration in seawater. practical The HgCl 2 solution was made as concentrated as (0.2 M.) to maximize the sensitivity of the method. Millipore (0.45 pm.) filters were used for the filtrations. Sulfite standards were prepared with sodium sulfite in distilled water treated as for the sulfide method. solutions were standardized iodometrically. The The standard error was + 1 pM. at the 95% confidence level. The absor- bances of the samples were then compared to the standard curves which had been fitted by linear regression. The detection limit was 1 1M. sulfite. Thiosulfate, Trithionate, & Tetrathionate The methods of T. Mizoguchi and T. Okabe (1975) were followed for thiosulfate, trithionate, and tetrathionate measurements. The three procedures are based on the reac- tions of these species with cyanide under different conditions (Table 2.5). The first procedure (A) is cupric ion- catalyzed cyanolysis of thiosulfate at a pH of 4.5. The second method (B) is the cyanolysis of tetrathionate at high pH, in the presence of acetone followed by cupric ion-catalyzed cyanolysis of thiosulfate. The last method (C) h 5 -27Table 2.5 Procedure Reaction 2- + CN -Cu 2 - 2SCN +SO 2- (1) S 0 (2) -Cu S O 2- + 2CN + 20H- 2 3 Equivalents of SCN expected (moles) 3 2+ 2SCN + SO 2- SA= (S2 0 3 S B= (S2 0 3 22(S + SO 4 2-+ H20 (1) S203 S23 2. (3) S3062 (1) + CN -Cu + SCN + SO 23 2- )+ B 20 ) 4 6 2- 2- + CN + 20H = SCN + SO 3 2- +SO 2+ -3 ) 3 -Cu + 2CN 4 6- + 20H -3 2SCN +SO 2+ 2-Cu 2S O + CN SCN + SO (2) S06 2- 2- 2+ 2 2- 2- 4 2- + 2- +SO 4 2- 2- Sc = H20 Sc = + H 0 2 (203 2-)+ (S 3 0 6 22) + 2(S4062 3 6 4 6 l~li .~~-ill__/l__I___I__l II~C^^i~_l_ _PI_____~_1_~ _ I~.._^ ~.-C. .-.rl~i~-X-i~ i._i~I-I.. -28- involves the cyanolysis of trithionate and tetrathionate in a boiling water bath at high pH, followed by cupric ioncatalyzed cyanolysis of thiosulfate. Cadmium acetate was added to the samples (Figure 2.4) to eliminate interferences from sulfide and sulfite (B. Sorbo, 1957 and P.J. Urban, 1961). Pre combusted approxi- mately 1 pm. glass fiber filters were used to remove the precipitate. The samples, where noted, were maintained at 190 C. in a thermostatically controlled water bath. The absorbances were measured at 460 nm. in a water cooled (190C) 1 cm. quartz cell. Cupric sulfate was substituted for cupric chloride in the above reactions (A, B, & C) when the cupric chloride solution had been used up on board ship. Standard curves for method A were prepared using a previously standardized 0.102 N. sodium thiosulfate solution (Vogel, 1961). The solution was made up following the sul- fide procedure with Na2S203 and standardized with potassium iodate. The error obtained from replicate standard curves fitted linearly was + 15 pmoles/liter at the 95% confidence level. Thiocyanate standards that had been previously compared to thiosulfate standards were used for methods B and C. The standards plotted in a parabolic curve (Figure 2.5), with a minimum of absorbance around 15 pmoles/liter of thiocyanate. Substitution of a less concentrated solution of cupric x -.rlilP1-1 -----il---1-I-*LIPII~------LIX~ -29- Figure 2.4 Thiosulfate, Tetrathionate, and Trithionate Analytical Procedure *iill .~ _.._ .~L_ ~........ _~._ CLX r~Y*III1IIYllllj^_~ ------YIU1III~-I I~C--III. -- .I~LI -30Figure 2.4 150 il. acetate I 10 ml. sample to 19 0 C cool III 20 ml. sample add 3 ml. acetate buffer pH adjusted to 9.3 w/ IM. ammonia water ad -3ml mix add 0.2 ml. KCN mix I I B 10 ml. 10 ml. add 3 ml. acetone add 0.4 ml. KCN cool to 190C mix, boil for 30 min., cool to 190 C 0.4 ml. KCN added mix & wait 20 min. add 0.3 ml. CuC12 or CuSO 4 mix add 0.3 ml. CuC12 or CuSO 4 mix add 1.5 ml. Fe (NO3 ) 3 mix measure Abs. at 460 nm. in water cooled (190C) cell ~..... --r-...r ;-;-- lr- r~l^l-ixc~ -ii-r=iI-* u~-iurax~--^.rs~ -- '-i'p~--i- u--- -Ir~;icsl uurPnlXL+slll---I~~'LI-~i~ -31- Figure 2.5 Thiocyanate, versus Absorbance, Method C, (CuSO4 ) 4 50 l 40 S @0@ MII I- 30 20 - S S. 0 10 550 ABSORBANCE 600 6 4 -33sulfate for the cupric chloride in the procedure increased the standard error (Table 2.6). The parabolic shape of the standard curves in these procedures (B and C) may be due to an interference caused by the seawater medium since this phenomena is not observed in distilled water. In general, errors on all the below procedures might have been further minimized if initial manipulation of the samples had been carried out under a nitrogen atmosphere. This, however, was not possible on board ship due to a shortage of nitrogen. Table 2.6 Std. error at 95% conf. (2a) pM. thiocyanate (CuCl2 ) + " (CuSO4 ) + 10 Method C (CuCl2 ) + 4.5 " (CuSO4 ) + 4.7 Method B 7 ___=~I~ I_ 1 _-I-_YI._U--~L---IC~II^ -~--- ~_~_I~_~ __ ___.__II~YIX111~11_1C-^~ .-. -34Table 2.7 Summary of Method Std. Errors Std. error at 95% confidence iM./liter Method Sulfide (1-3 f" _L4) + 0.05 (3-40 '_4.) + 0.9 (40-250 PM + 29 Sulfate + 0.2% Iodine Reactive Species + Sulfite + 1 S203(A) 65 + 15 SCN (B, CuC1 2 ) + 7 SCN (B, CuSO 4 ) + 10 SCN (C, CuCl 2 ) + 4.5 SCN (C, CuSO 4 ) + 4.7 --. I~ -I--IIII~L_-EI--II. ^^__ Il---l~b-.I I-I~-~~C-I~XIII)_.-.__. -35REAGENTS Sulfide: N,N- Dimethyl-p-phenylene-diamine Sulfate; [NH 2 C 6 H 4 N(CH 3 )2 2 H 2 SO 4 , Eastman Kodak Co., No. 1333. Ferric Chloride; FeCl 3 . 6H20, A.C.S. Reagent grade, Matheson Coleman & Bell. 6N. Hydrochloric Acid; A.C.S. Reagent grade, Fisher Scientific Co.. Sodium Sulfide; Na2S-9H20, A.C.S. Reagent grade, Mallinckrodt, Inc.. Iodine Reactive Species: 0.1 N. Iodine; A.C.S. Reagent grade, J.T. Baker Chem. Co., (standardize daily). Starch soln. - Make a paste of 1.0g. of Soluble Starch; A.C.S. Reagent grade, Merck and Co., Inc., with a little distilled water, and pour the paste with constant stirring, into 100 ml. of boiling distilled water, and boil for one minute (A.I. Vogel, 1961). _;r.^-~-- .~-^--XI^-~xl--~nr-^-_m.r.rir.nns--.X.1DLX*-~ ~L -I~-*~-I~I~L~--~-IIPXI~PU_yCI r^l--I-Y---IL--- -360.1 N. Sodium Thiosulfate; Na 2 S2 0 3 .5H2 0 ) A.C.S. Reagent grade, Mallinckrodt, Inc.. Potassium Iodate; KIO 3 , A.C.S. Reagent grade, Allied Chem., Specialty Chemicals Division. Potassium Iodide; KI, A.C.S. Reagent grade, J.T. Baker Chem. Co.. 2N. Sulfuric Acid, H2S04, A.C.S. Reagent grade, Mallinckrodt, Inc.. Sulfite: 3M. Phosphoric Acid; H 3PO 4 , A.C.S. Reagent grade, 85%, Fisher Scientific Co.. 0.2 M. Mercuric Chloride; HgC1 2 , A.C.S. Reagent grade, Mallinckrodt, Inc.. 0.6% Sulfamic Acid; NH 2 SO 2OH, Assay 99.90-100.10%, G. Frederick Smith Chemical Co (Prepare daily). 0.2% Formaldehyde; HCHO, A.C.S. Reagent grade, Approximately 37%, Mallinckrodt, Inc., (Prepare daily). IM. Sodium Acetate-Acetic Acid buffer; Na2C2H302 3H20, A.C.S. Reagent grade, Matheson Coleman and Bell. Acetic Acid Glacial; CH 3 COOH, A.C.S. Reagent Grade, 99.7%, Fisher Scientific Co.. ..(-1~-~YII~____~_.-^ ~-~I~III-^.~L-I._1II-C--ULII~..i~l-~il..~~~~ 1I l~l~ I~-~~-IIICII ^I~s~ll~*-_I~1L-----I--^11111 -.. ^__i-1L.I - ilY--~~ ----- -37Pararosaniline Chloride; C 9H18ClN 3 , (0.2% + 0.03 in 1 M. hydrochloric acid), Eastman Kodak Co., A 14051. P.C. concentration (Figure 2.6) Pararosaniline Chloride (P.C.), 0.2% stock solution 1 ml. Dilute to 100 ml. with distilled water 5 ml. Add 5 ml. acetateacetic acid buffer. Dilute to 50 ml. with distilled HO Wait 1 hour, measure absorbance at 540 nm. r I~-PPsl~LI -- IYL1~-.~ --- r~l~-lx. l-.-.-I.~-. I -nr--~x---- ---- UC- --U~L~~ -~-Ili~~ll~CllTI~ 11I.1~~~ *~L----~~iL-ellrii3-_.__~-~~L.rr.l~..^-----.--~ *--~---lyn~l -38The actual pararosaline concentration as a percentage of the nominal concentration is determined by the formula: (1) % P.C. = Abs x k grams of dye taken to allow for correction of P.C. (lA) % P.C. = (.458) , where k = 21.3 in reagent. (21.3) = 97.5% P.C. (.1) Procedure B (Scaringelli, et al., 1967) was used because this method covered a wider range of sulfite concentrations. PRA reagent for method B is prepared by adding 200 ml. of 3 M. phosphoric acid to 20.5 ml. stock P.C. and diluting to 250 ml. with distilled water (PRA reagent). An additional 0.2 ml. of stock P.C. was added for each one percent that the stock P.C. assays below 100% (0.2 ml. x 2.5% = 0.5 ml.). Sodium Sulfite Anhydrous; Na 2 SO 3, A.C.S. Reagent grade, Merck and Co., Inc.. Thiosulfate, Trithionate, and Tetrathionate: 0.75 M. Potassium Cyanide; KCN, A.C.S. Reagent grade, Matheson Coleman and Bell. 0.3 M. Cupric Chloride; CuCl2*2H20 , Analytical Reagent grade, Mallinckrodt, Inc. OR 0.2 M. Cupric Sulfate; CuSO 4 .5H 2 0, A.C.S. Reagent grade, J.T. Baker Chem. Co.. /~___ X^__I~____PY___U_____LI~_~\ I41~-rllYIIIII~---L1 e -390.2 M. Cadmium Acetate; (CH3 CO 2 )2 Cd2H2O, Reagent grade, Matheson Coleman and Bell. 2 M. Sodium Acetate - Acetic Acid buffer; (see above). 1M.Ammonium Hydroxide; A.C.S. Reagent grade, Assay 28.0-30.0% NH 3 , Fisher Scientific Co. 1.5 M. Ferric Nitrate - Perchloric Acid Reagent303 g. of Ferric Nitrate; Fe(NO 3 )3 9H2 0, A.C.S. Reagent grade, Mallinckrodt, Inc. was dissolved in a small volume of distilled water containing 186 ml. of conc. Perchloric Acid; HC104, A.C.S. Reagent grade, 70 wt.%, G. Frederick Smith Chemical Co., and diluted to 500 ml. with distilled water. 0.1 N. Thiosulfate; (see above). Potassium Thiocyanate; KCNS, A.C.S. Reagent grade, Fisher Scientific Co.. ^I-iipl~l----l--l-li~i~. . -~~111~1 111_ -40- CHAPTER 3 RESULTS AND DISCUSSION ~-~-_L .~IP(-.Y-X-~FIP..XI-~-I^--I--I----Yl~ L~.~ n--I~^-^*PII-~--YI~-~ -C~~~---III--IWI -41Sulfur species in the Galapagos hydrothermal waters show linear trends versus temperature in the data (passing through the ambient sea water temperature and composition) which were interpreted as dilution lines. Dilution of the concentration of the species was due to a combination of mixing below the seafloor, in the vents, and during sampling with ambient sea water (J. B. Corliss, et al., 1979 and J. M. Edmond, et al., 1979a,b). Problems with the sampling system leaking led to further dilution (0-89% hydrothermal water collected) and possible oxidation of the reduced sulfur species present in the samples. Sulfide concentrations in the vents increased with increasing temperature (Table 3.1, Figure 3.1). East of Eden had the highest gradient (2.6 x 1012 mol/yr) with Mussel Beds changing the least (Table 3.2). These trends indicated that hydrogen sulfide was being produced in the hydrothermal system, either by reduction of seawater sulfate, leaching sulfide from the basalt or primary hydrogen sulfide from magmatic sources (Bonatti, 1975). Sulfide concentration versus oxygen concentration (Figure 3.2)showed a decreasing trend. Oxygen was absent in the samples with hydrogen sulfide concentrations above 120 pM, which differs substantially from data previously published (Edmond, 1979b) where oxygen disappeared at approximately 50 M sulfide. This difference is due to the entrainment of oxygenated __\_YIY__^C___1__L_)- li--~i~~-I.I-._Y1I_ I-r~Y LIL~-X -..~~II -~Y~-I-I*LL. -42- Table 3.1 Concentrations of Sulfide, Sulfite, Thiosulfate, Trithionate, Tetrathionate, Iodine Reactive Species, Oxygen, and Silica for the Galapagos Vents -43Table 3.1 Dive # Vent #I pM. H 2!S iP1. S203 PA - Si 02 Bottle #2 1M. 2- SO 3 2- PM. 12 peq. SOM4 VM. 2- 2- s 02- S 06 A. 898 1 207 275 84 1.4 1 -1 336 11 -5 898 1 204 259 114 1.6 9 -1 312 9 -5 898 1 103 402 48 10.5 2 0 581 898 1 202 400 60 16.7 2 1 589 898 1 203 457 36 19.7 8 1 581 898 1 206 201 106 7 -1 899 2 105 619 0 147.0 5 899 2 106 596 0 144.0 899 2 107 587 0 165.0 899 2 101 513 4 899 2 109 550 0 145.0 899 2 113 479 0 899 2 112 395 899 2 102 303 900 2 202 854 900 2 206 846 12 1 12 -3 12 -8 450 10 -3 1 216 12 -4 11 1 277 6 7 18 1 314 5 -6 9 0 335 27 0 220 1 67.0 8 1 196 10 -6 19 44.0 7 1 167 11 -6 58 49.0 6 1 151 11 2 0 177.0 14 1 453 7 -4 0 285.0 18 1 510 0.3 75.0 28.16 28.24 27.88 27.42 9 0 4g Cr~ -44Table 3.1 Dive # Vent # Si 02 H S Bottle #2 PM. PM. PA. (contId.) 2- SO 3 2,_.3 12 3 peq. s 0622-- 2-. PM. 8 -5 8 -7 379 7 -7 3 188 9 -7 3 183 10 -6 4 198 369 900 2 103 625 0 190.0 14 0 900 2 207 348 34 93.0 14 2 900 2 204 608 0 162.0 17 0 900 2 203 322 54 98.0 13 900 2 205 279 66 80,0 11 901 3 109 161 117 0.5 901 3 102 161 119 0.4 288 901 3 107 160 115 0,9 288 901 3 112 167 109 0,7 300 901 3 106 336 0 142.0 3 316 901 3 105 360 0 182.0 -7 406 902 1 204 605 0 31.5 15 77 902 1 203 626 0 35.8 21 98 902 1 104 490 43 21,9 902 1 207 575 0 31.5 73 13 28.30 27.78 18 -10 8 -7 5 2 12 -6 6 0 -45- Table 3.1 (cont'd.) Si 02 H2S S 20 3 SO3 PM. pM. 3 12 peq. 2- SO 4 2-s06 PM. pA. S 06 2- uM. Vent # Bottle #2 PM. PM. PM. 902 1 202 486 13 22.4 29 0 -20 1 902 1 205 486 10 19.1 14 0 82 8 902 1 103 443 55 12.9 0 1 16 15 903 2 102 558 0 137.0 11 1 143 10 2 903 2 107 837 0 140.0 5 1 384 9 5 903 2 106 927 0 221.0 4 1 633 13 -5 903 2 101 636 0 138.0 5 1 45 12 -4 903 2 109 724 0 169.0 5 1 294 903 2 302 420 26 118.0 6 1 904 1 205 588 0 28.1 28 -1 132 904 1 207 561 3 24.5 28 0 120 904 1 204 507 15 20.7 25 0 904 1 202 595 0 33.0 25 0 904 1 303 574 11 28.6 27 905 2 113 1007 0 336.0 18 Dive # 27.47 27.78 -1 2 -6 12 2 11 0 0 -1 0 0 28.02 2 0 140 27.84 2 -2 0 112 27.87 1 -1 1 446 27.14 4 -1 62 59 27.81 -46- Si Table 3.1 (cont'd.) 22HS 0 S03 S yM. 3 PM. PA. ,,A. 3 12 -peq. Bottle #2 PM. 905 106 238 33 31.9 15 905 112 372 28 70.0 15 905 101 627 0 190.0 15 905 109 596 0 148.0 15 1 184 905 304 343 317 7 1 -11 165 120 Dive # Vent # 0 I.: Mussel Beds; 2: 1. Vent #: 2. Bottle #: 3. Iodine Reactive Species 1 : A; 2 : B; 3 : H; 2- 2- s 2S0 60 -5 233 0 0.0 Rose Garden; 3: SOM. VM. East of Eden (e.g. 105 --A 5). 27.64 5 1 -3 28.62 0 -I~LUIEPr~s~- -47- Table 3.2 Gradients and Fluxes for Sulfide and Sulfate -~r~-rULll^_nyulr~ -48Table 3.2 SO Range 0.0-336 moles ) Gradient (Cal. Cal. (1) 5.14 x 10 (2) 1.83 x 10 (3) 5.24 x 10-8 5.24 x i0 Flux moles ) . M. 11 (1) 2.6 x 1011 (2) 9.2 x 10 2- 27.14-28.62 mM. Iodine Reactive Species -20-633 (1) -1.06 x 10-7 -1.06 x i0 - 12 -5.3 x 1012 (3) 2.6 x 1012 peq. -8 6.08 x 10 (1A) 3.27 x 10-8 (2) 3.65 x 10 (3) 3.13 x 10-8 12 (1) 3.04 x 1012 (LA)1.63 x 10 (2) 1.83 x 1012 (3) 1.56 x 1012 For the above calculations, 5 x 1019 cal/yr. was taken as the global hydrothermal heat transport from accreting plate boundaries. (1) Mussel Beds (2) Rose Garden (3) East of Eden , (lA) Mussel Beds, Dive 898 _I--_--- I _ ..I~IIIIIIYYCI~- -49- Symbols Used in Figures in Chap. 3 O - Mussel Beds + - East of Eden A - Rose Garden _ -PLIC-I^~TYI~IP~I~I~-Y) 1.I~1I~--_1( 11III~-~.I^L----Llr*rillllX- ~Li_--il__i( - ...~.i_^ll. .---IPI_~- . . -50- Figure 3.1 Sulfide versus Silica S-51- ,wI a *6* M In + UA NI 100 iA Oo (")O AA w 200 A A A $00 %V 300 400 SILICA pM. 900 1000 1100 I~-*~--. lil__~llll*____1_____~ _IPI_; 1)~ -52- Figure 3.2 Sulfide versus Oxygen -l~--si-l. .~-1X~ -53- 350 300~ 300 A 2501 200 u.u Q. ,=6 150 100 50 0 c o | A I 10 20 OXYGEN pme 80 Ip I % 90 100 o110o120 _ I^lr~_i__l~____ I ~ll----- - IIIICIII1* ~ LYIIII--PIXI~I-~~--rl -54ambient water during sampling with the leaky samplers and the relatively slow oxidation of sulfide. (Sulfide oxidation is inhibited at pH values below 7, presumably because of the protonation of the HS molecule, Chen and Morris, 1972). Sulfate decreased uniformly in all vent fields (Figure 3.3, Tables 3.1, 3.2). Sulfite was constant with temperature at about 1 pM (the detection limit), (Figure 3.4), though in East of Eden and Rose Garden some of the concentrations at low temperature were above 1 M sulfite. Thiosulfate, trithionate, and tetrathionate were also constant with temperature within the limit of resolution (Figure 3.5, Table 3.1). Cyanolysis of any elemental sulfur present (only particles <1 pm as the samples were filtered) should interfere positively to some extent in the tetrathionate method and to a greater degree in the trithionate procedure (Nelson, et al., 1977), though this interference has not been quantified. Species reduced by iodine showed a general increase with temperature (Figure 3.6). The vent fields differed in their gradients (Table 3.2) with Mussel Beds exhibiting two different trends associated with two different days of sampling. In water with a neutral or acidic pH (hydrothermal waters--pH 6 to 7) only sulfide, sulfite, s _I_ ~_I~ I~--~------------"III~arr~--Ll~iPIIY IZL -55- Figure 3.3 Sulfate versus Silica i-'-ivl'lir~ -x~--~1li1--~ - I~I ~I--- -- -i-(r~_ -56- 29 E I- 28 -U 27 r r I 200 300 400 I SILICA I I pM. 800 900 1000 1100 1. ~-T l)i_ ICI-X----.I.- -57- Figure 3.4 Sulfite versus Silica -58- 4 S U L F I AA A E iPMo &A Ab A.dM 'AI i tj Ar J 1 I v - MM A&VV'N m T v l i - - I -1 -2L 100 SIL.ICA pM. 200 300 400 500 600 700 800 900 1000 1100 ____l*l__lj~ll*___C__IP_-C.~ -59- Figure 3.5 Thiosulfate versus Silica ~--I---X~-L - LLI_(-LYL_. - liUCI Ill_~l -~X L~l L-LI1 -60- 30 20 AA 10 aL I- OAA A I, :) tn U6 2 zI- A AA 0 -10 I-.I - : I 200 o . . "/ • 300 400 V I ! ! • 500 600 SILICA pM. 700 I 800 900 1000 1----L1-~ IPIPrrPP~-~1~~ -rilr)~a3 ~lrr.~--~ Il~-------~_izr ~yll... --~r.~ p~__.. -61- Figure 3.6 Iodine Reactive Species versus Silica 4 4 0 46 -62- U) 600 400 0 0ILl w11 400 U z~d~ 2k 200 m- 300 A A * + k A A A % 100 -100 A 200 300 '400 500 SILICA 600 700 800 pM. 900 1000 1100 I~-~L--^II L;~ ___ZILII^__*XIll___-*II~ -63and thiosulfate should be expected to be reactive with iodine. The concentrations of these sulfur species leaves a large amount of reacted iodine unaccounted for (Figure 3.7). A possible explanation for this is suggested by experiments by Nelson, et al. (1977); colloidal sulfur suspensions stabilized by thiosulfate or polythionates may lead to iodine reactivity, unlike other sulfur suspensions. This reactivity may be due to a surface charge mechanism resulting from slow hydrolysis or oxidation upon the surface of the sulfur crystals, whereby charge sulfoxy groups react. The hypothesis that these unaccounted for iodine reactive species are in fact colloidal sulfur is further supported by the observation that the major component of suspended material filtered in situ from the hot springs is native sulfur (Corliss, unpublished data). Polysulfides, while also iodine reactive, were not measured since their presence is unlikely due to the low pH conditions present in the hydrothermal solution. In a profile of the overlying waters 135 and 139), (stations 86, reduced sulfur species were not found. The sulfur chemistry of the Galapagos hydrothermal vents suggest that hydrogen sulfide and possibly elemental sulfur are the principal reduced sulfur species. thiosulfate, and polythionates are observed in low Sulfite, ~_rsr~-iYr~ -- a-~-r~----r*li-----L _r~.~....~l.itn~l^ir-~-rr~- -64- Figure 3.7 Iodine Reactive Species Minus Sulfide, 2x Thiosulfate, and Sulfite (12 ) versus Silica -65- 500 400- 300 0) + AA 2001+ C4 A A 100- A A #9 4 SA * W A * A*P AQ A9A- - 100oo 100 200 300 400 500 SILICA 600 700 pM. 800 900 1000 1100 ~___j LI_l_____n_ I__IC_____J_~_I___~IUL___I_ -66concentrations implying that sulfur is not oxidized beyond the zero valence state to any significant extent. The flux of sulfide from the hydrothermal system is large, 2.6 x 1011 2.6 x 1012 mol/yr, which is consistent with previous observations (Edmond, et al., 1979b). Sulfate is reduced in the vents (flux = -5.3 x 1012 mol/yr) suggesting that this is an important sink in the oceanic sulfate budget (Edmond, et al., 1979a). The observed concentrations of sulfur species in the hydrothermal vent waters suggest that elemental sulfur and possibly polysulfides should be examined in more detail as the measurement of these species will give additional insight into the sulfur chemistry of seafloor hydrothermal systems. -67- References --lr r^r --r~--rrP-Nr~ilX1*lyr~ IQIL~LI~LICli LI1^Y---~ ~ -68- References Bonatti, E. (1975). Centers. Metallogenesis at Oceanic Spreading Annual Review of Earth and Planetary Sciences,3, 401-431. Brewer, P.G. (1975). Minor Elements in Seawater. In J.P. Riley and G. Skirrow (eds.), Chemical Oceanography, (Vol. 1, 2nd ed.), Academic Press, London, 415-496. Budd, M.S. and H.A. Bewick (1952). Photometric Determination of Sulfide and Reducible Sulfur in Alkalies. Analytical Chemistry, 24, 1536-1540. Chen, K.Y. and J.C. Morris (1972). Kinetics of Oxidation of Aqueous Sulfide by 02. Environmental Science and Technology, 6, 529-537. Cline, J.D. (1969). Spectrophotometric Determination of Hydrogen Sulfide in Natural Waters. Limnology and Oceanography, 14, 454-458. Cline, J.D. and F.A. Richards (1969). Oxygenation of Hydrogen Sulfide in Seawater at Constant Salinity, Temperature and pH. Environmental Science and Technology, 3, 838-843. Cline, J.D. and F.A. Richards (1972). Oxygen Deficient Conditions and Nitrate Reduction in the Eastern Tropical North Pacific Ocean. Oceanography, 17, 885-900. Limnology and ~L~---~. IY*lle;-~~-IVI ______/_LI~_L___PIYYIC_ -69Corliss, J.B., J. Dymond, L.I. Gordon, J.M. Edmond, R.P. von Herzen, R.D. Ballard, K. Green, D. Williams, A. Bainbridge, K. Crane, and T.H. van Andel (1979). Submarine Thermal Springs on the Galapagos Rift. Science, 203, 1073-1083. Edmond, J.M., C. Measures, R.E. McDuff, L.H. Chan, B. Grant, L.I. Gordon, and J.B. Corliss (1979a). Ridge- crest Hydrothermal Activity and the Balances of the Major and Minor Elements in the Ocean: Galapagos Data. the Earth and Planetary Science Letters, (in press). Edmond, J.M., C. Measures, B. Mangum, B. Grant, F.R. Sclater, R. Collier, A. Hudson, L.I. Gordon, and J.B. Corliss (1979b). On the Formation of MetalEarth and Plane- Rich Deposits at Ridge Crests. tary Science Letters, (in press). Goldhaber, M.B. and I.R. Kaplan (1974). The Sulfur Cycle. In E.D. Goldberg (ed.), The Sea, (Vol. 5) John Wiley and Sons, New York, 569-655. Humphris, S.E. and G. Thompson (1978). Hydrothermal Alteration of Oceanic Basalts by Seawater. Geochimica et Cosmochimica, 42, 107-125. Luther, G.W. and A.L. Meyerson (1975). Polarographic Analy- sis of Sulfate Ion in Seawater. Chemistry, 47, 2058-2059. Analytical LI LII _IIC______Ll_____lr___III1I_--L- -70Mizoguchi, T. and T. Okabe (1975). The Chemical Behavior of Low Balance Sulfur Compounds. IX. Photometric Determination of Thiosulfate, Trithionate and Tetrathionate in Mixture. Bulletin of the Chemical Society of Japan, 48, 1799-1805. Mottl, M.J., H.D. Holland, and R.F. Corr (1979). Chemical Exchange During Hydrothermal Alteration of Basalt by Seawater-II. Experimental Results for Fe, Mn, and Sulfur Species. Geochimica et Cosmochimica Acta, 43, 869-884. Nelson, M.B., J.A. Davis, III, M.M. Benjamin, and J.O. Leckie (1977). The Role of Iron Sulfides in Controlling Trace Heavy Metals in Anaerobic Sediments: Oxidative Dissolution of Ferrous Monosulfides and the Behavior of Associated Trace Metals. Sorbo, B. (1957). NTIS Report AD-A049712. A Colorimetric Method for the Determina- tion of Thiosulfate. Biochimica et Biophysica Acta, 23, 412-416. Urban, P.J. (1961). Colorimetry of Sulfur Anions, Part 1, An Improved Colorimetric Method for the Determination of Thiosulfate. Chemie, 179, 415-422. Zeitschrift fuer Analytische _ i*.~-yllllll^~--II*-- 1~-1sl~ 11(L11~ -- ~-1-1-_1111 -----~C.~- 1-LI~Cl~-. 11~11~3 1L^.-1__I-L 9-I~LIIY.-P^I~-l^ ---ll~ ~ _I~-1 __i -71Vogel, A.I. (1961). A Textbook of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis (third ed.). New York, 1216 pp. John Wiley and Sons Inc.,