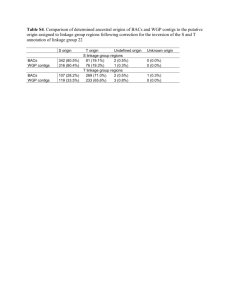
Document 10614145
advertisement

Functional annotation of Transcriptomes assembled de-novo from RNA-Seq data using
Random Walk with Restart
12
Jose, Adarsh ; Yandeau-Nelson,
1 Department
1
Marna ;
J. Nikolau,
1
Basil
of Biochemistry, Biophysics & Molecular Biology, Iowa State University; 2 Bioinformatics & Computational Biology Graduate Program, Iowa State University
Introduction: We formulate the problem of identifying and
prioritizing homolog contigs of known genes/gene families in
de novo-assembled transcriptomes as an instance of the
Random Walking with Restart problem on a cross-organism
sequence similarity network. The algorithm uses shared
neighborhoods in cross organism sequence similarity networks
to identify homolog contigs and assign scores to rank them.
Background: The advent of ultra-high throughput sequencing
technologies has resulted in the accumulation of terabytes of
short read transcript sequence data. Currently, there are a
number of very efficient algorithms (eg. Trinity2) that can
assemble these short reads into contiguous fragments of
consensus sequences (contigs).
A standard approach for assigning function to new sequences is
based on the assumption that sequences that share sequence
homology share functionality. Upon a BLAST analysis, contig
function is assigned based on the functional annotation of the
most significant hit. However, the shortness of the reads (100
bp) will result in fragmented contigs (Figure 1), which
introduces uncertainty about the true parent transcript of an
assembled contig.
Random Walk with Restart (RWR): The random walk (RW)
on a graph is defined as the iterative transition from a given
node to its randomly selected neighbor. The random walk
with restart returns to the start node of the walk at each step
with a user-defined probability. Formally, the RWR is
defined as:
α ≥ 0.30 & e-value < 0.05
α < 0.30 & e-value < 0.05
where W- column-normalized adjacency matrix of the graph
,pt(i) - Probability of being at node i at step t; r – probability
of return to start node at each step.
C
0.6
0.5
0.8
D
A
B
C
D
E
F
E
0.4
0.8
0.7
A
0
0.8
0.8
0.7
0.4
0.5
B
0.8
0
0
0
0
0.7
C
0.8
0
0
0.6
0.5
0
D
0.7
0
0.6
0
0
0
E
0
0
0.5
0
0
0
F
0.5
0.7
0
0
0
0
Run RWR:
po (x)= {1/|Query|, if x ϵ {Query}, otherwise 0}
Contigs from Corn Silks
B
KCS gene family in
Arabidopsis thaliana
0.7
Most significant hit
α ≥ 0.30 & e-value < 0.05
α < 0.30 & e-value < 0.05
F
0.72
The long hits are
ACC-2 genes from
Arabidopsis
C
0.60
0.6
0.5
0.8
D
0.40
E
0.4
0.8
0.7
A
0.61
B
0.5
0.7
F
0.60
Figure 2: An illustration of the RWR algorithm. Note that A-B and A-C
has same α score (0.8), but different RWR score. C has higher
score(0.72) compared to B(0.61) as it shares two neighbors(D and E)
with A and B only shares one neighbor(F) with A.
FLOWCHART
Gene models
from source
organism
Denovo- Assembled
Set of Contigs
Sequence Similarity Network and Random Walk with Restart:
• Sharing domains associated with characteristic functions
between genes and contigs translate to sharing multiple
neighbors in a sequence similarity network.
• The identification of homologs of a query gene reduces to
finding the nearest two-step neighbors in an all-versus-all
sequence similarity network.
• The problem of identifying and ranking homologs between two
phylogenetically related organisms , therefore, can be defined as
an application of the graph theoretical concept of Random
Walking with Restart3 where the cross-organism sequence
similarity network is defined by nodes (sequences) and edge
weights (measure of sequence similarity based on BLAST
scores).
Figure 3(a): Only edges with e-value <
The are shown in the graph. Even though,
0.05
all sequences have significant hits, only
some share multiple neighbors (multiple
dark edges to the query family) with each
other.
ILLUSTRATION
0.5
• A high BLAST score, therefore, is a necessary but not
sufficient indicator of true functional homology as the
fragmented nature of the contigs can result in “many-to-many”
mapping between genes and contigs.
• True homolog sequences, in addition to having high sequence
similarity, share functionally conserved domains with
several genes in its gene-family, even across organisms that
are close in the phylogenetic tree.1
• This facilitates use of well annotated genomes (e.g.
Arabidopsis thaliana) to annotate newly sequenced
transcriptomes.
Contigs from Corn Silks
KCS gene family in
arabidopsis thaliana
A
Figure 1: Example of fragmented contigs - The homologs of Acetyl
CoA Carboxylase (AT1G36160.1) from Arabidopsis thaliana visualized
on the NCBI BLAST server. The contigs are obtained from
transcriptome assembled de-novo from Corn (B-73) Silks. Contigs of
varying bit score and coverage show significant hits. The Contigs were
assembled using Trinity Transcriptome Assembler2
Homologs of Keto-acyl CoA Synthase (KCS) gene family from Arabidopsis
thaliana among De-novo Assembled mRNA Contigs from Corn Silks
All vs All BLAST &
Estimate Similarity α(i,j)
l: similar bases,
a: length of query sequence
b: length of target sequence
Graph G – {V,E};
Vϵ{Sequences},E ϵ { E(X,Y) = α(X,Y);
X,Y ϵ V; e_value(X,Y) < 0.05}
Query Gene / Gene Family
Initialize parameters and run RWR
Sort Homologs by their pt(Contig) score
normalized by max score
Figure 3(b): Result of the algorithm on the Graph in Figure
3. The scores are shown in the nodes. The true homologs get
high scores compared to weaker hits. (Note that this network is a
subset of the whole Graph and is for illustration purpose only. The score
difference between true homologs and others becomes more pronounced when
the entire graph is used.)
Conclusions:
• True homolog sequences, in addition to having high
sequence similarity, are known to share functionally
conserved domains with several genes in its gene-family,
even across organisms which are close in the
phylogenetic tree.
• We developed an algorithm which uses this property to
prioritize candidate homologs in denovo-assembled
contigs.
• We are currently testing the algorithm’s ability to identify
true homologs and the effects of different parameters and
network size on its efficiency.
References:
1.Song N, Joseph JM, Davis GB, & Durand D (2008) Sequence similarity
network reveals common ancestry of multidomain proteins. PLoS Comput
Biol 4(4):e1000063.
2.Grabherr MG, et al. (2011) Full-length transcriptome assembly from RNASeq data without a reference genome. Nature biotechnology 29(7):644-652.
3.H. Tong CF, J.-Y. Pan (2006) Fast random walk with restart and its
applications. Proceedings of the 6th International Conference on Data Mining,
IEEE Press:p. 613-622.