Document 10446004

MSA Comparative Crystal Chemistry
Chapter 7
The Analysis of Harmonic Displacement Factors
R.T. Downs
INTRODUCTION
In my role as crystallographic editor for American Mineralogist and The Canadian
Mineralogist, I examine many papers that include crystal structure refinement data and discussions about this data. It is clear to me that understanding and working with the displacement parameters is a challenge to many researchers. The purpose of this chapter is to clearly define the meaning of these parameters and the mathematics needed to interpret them. Throughout this chapter, I will provide illustrative examples from the structure of quartz refined by Kihara (1990) at a variety of temperatures.
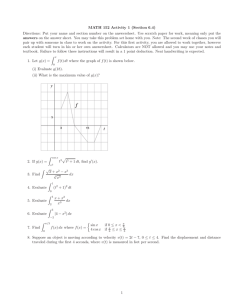
Figure 1.
An image of the displacement ellipsoids for a Si quartz, SiO
2
2
O
7
group in α -
, at 298 K and 838 K (Kihara, 1990). The ellipsoids, as drawn, enclose 99.0 % of the probability density.
It is well established that an atom in a crystal vibrates about its equilibrium position.
This vibration can be attributed to thermal and zero-point energy. For example, in Figure
1 the anisotropic displacement parameters are drawn for the Si and O atoms in quartz at
298 K and 838 K. The displacements are significant, with maximum amplitudes for O of
0.138 Å, and 0.259 Å, at 298 K and 838 K, respectively, in the direction perpendicular to the Si-Si vector. Therefore, in a crystal structure refinement, it is imperative not only to find the mean position of an atom, but also to describe the region in space where there is a high likelihood of finding it. This region of space can be mathematically defined with a probability distribution function (p.d.f.). If we assume harmonic restoring forces between the atoms, or in other words, that the forces between the atoms are quadratic and obey
Hooke's Law, then it can be shown that the p.d.f. can be represented by a Gaussian function (Willis and Pryor, 1975 p92). The topic that links atomic forces with thermal motion is called lattice dynamics and is not the subject of this chapter. For an introduction to the subject of lattice dynamics see Born and Huang (1954), Willis and
Pyror (1975), or Dove (1993). The work of, for instance, Pilati et al (1994), illustrates the use of lattice dynamics to compute the displacement parameters. However, from the
1
theory of lattice dynamics is known that the p.d.f should be bounded by the contours of constant energy that envelop an atom within a crystal structure (Figure 2a). Furthermore, it follows from density-functional theory that the p.d.f. should be oriented in such a way that the long axis of the ellipsoid points into directions of relatively lower electron density (Figure 2b). Other uncertainties can also be represented by this p.d.f., such as, for instance, substitutional and lattice defects or positional disorder (Hirshfeld, 1976;
Trueblood, 1978; Dunitz et al, 1988; Kunz and Armbruster, 1990; Downs et al, 1990).
Figure 2 . Maps of (a) energy contours and (b) procrystal electron density in the plane perpendicular to [210] centered on the O atom in β quartz at
891 K with the thermal ellipsoid of the O atom superimposed. The energy was computed with the SQLOO energy function constructed for silica by
Boisen and Gibbs (1993). The thermal ellipsoid is from Kihara (1990).
The SiO bonds are projected onto the plane. By theory, the ellipsoid should be parallel to the energy contours. This discrepancy may be due to an insufficient energy function and/or errors in the refined values of the thermal ellipsoid.
The Relation Between Displacement Factor and Probability Distribution Function
During a typical crystal structure refinement, the recorded intensities of an X-ray beam, diffracted from a large number of reciprocal-lattice vectors, s , [ s ]
D*
= [hkl] t , (for a review of the notation, see Boisen and Gibbs, 1990) is converted to a set of structure amplitudes, |F( s )|. This set of observed amplitudes is compared to a set of structure amplitudes calculated with the equation
F( s ) = n ∑ ƒ h j
( s ) e
2 π i r j
⋅ s
(1) where and r j
ƒ
ƒ between the measured and calculated amplitudes is then minimized by varying r j h j
( s n
). The value of r j j = 1
is the number of atoms in the unit cell,
represents the positional vector of the j th h j
( s ) is the hot atomic scattering factor,
atom in direct space. The difference
and
obtained from this minimization procedure represents the reported atomic coordinates for atom j .
2
The hot atomic scattering factor, ƒ h j
( s ), represents the effect that the various electron distributions, from the various atoms in the structure, have on X-ray scattering. There are two terms implicit to this factor. The first term is due to the electron distribution that is dominated by the contribution of core electrons. These electrons move so quickly in the regions around a stationary atom that, during the time of an experiment, they can be considered, by time averaging, to be represented by a constant electron distribution term.
The value of this term is dependent on the species of atoms that are present in the structure. The second term is attributed to the slower vibrational motion of the atom and its value is dependent on local interatomic forces, which, of course, vary from crystal to crystal and from position to position within the crystal.
It will now be shown that the hot atomic scattering factor is the product of the cold atomic scattering factor, (the electron distribution), with the Fourier transform of the p.d.f., (the vibrational motion of the atom), i.e.
ƒ h j
( s ) = ƒ c j
( s ) ℑ (P( r )).
Consider a small volume element, d V, containing P( r ) d V electrons, located at the end of the vector r (Figure 3). Then the path of an X-ray of wavelength λ , travelling from its source to the detector, passing through the end point of r , has increased by λ r ⋅ s over the path of an X-ray passing through the origin of the vector r (Lipson and Cochran, 1953, p
3-7). If and s
1 s
0
is a vector parallel to the direction from the X-ray source to the end point of r
is a vector parallel to the direction from the origin of length 1/ λ , then s is defined as s = s
0
s
1 r to the detector, both of
. The change in the phase of an X-ray passing through the end point of r over an X-ray passing through the origin is this increase in distance, multiplied by the rate of change of phase with distance, 2 π r ⋅ s (Feynman et al,
1977, chapter 29). The amplitude of the wave, scattered from a point source at r , is then proportional to P( r ) e
2 π i r ⋅ s d V, where the proportionality constant is the cold scattering factor, ƒ c
. The exponential term represents the decrease in intensity caused by a change in phase due to path difference. Since P( r ) is a continuous distribution, the amplitude of all the waves scattered by the distribution is found by the superposition principle to be
F( s )= ƒ c ∫
V
P( r ) e
2 π i r ⋅ s d V. (2)
3
Figure 3.
Scattering from two lattice points located at the origin, o , and the end point of vector r . The vector from the X-ray source is s
0
, and to the detector is s
1
.
The integral in Equation (2) is called the displacement factor or, sometimes, the temperature factor. If there are many atoms, and hence many electron density functions, all contributing to the X-ray scattering, then the overall amplitude is the superposition of all these distributions,
F( s ) = n
∑ j = 1
V
∫ ƒ c j
P j
( r r j
) e
2 π i r ⋅ s d V n
∑ j = 1
V
∫ ƒ r r j
) e
2 π i( r r j
) ⋅ s
e
2 π i r j
⋅ s
d V
≡ n
∑ j = 1
ƒ h j
( s ) e
2 π i r j
⋅ s , where n is the number of atoms in the unit cell. The hot atomic scattering factor for the j th atom is expressed as
ƒ h j
( s ) = ƒ c j
( s )
V
∫ P j
( r ) e
2 π i r ⋅ s d V = ƒ c j
( s ) ℑ (P( r )), where the displacement factor is the Fourier transform of the p.d.f.
ISOTROPIC DISPLACEMENT FACTOR
As the first approximation under the harmonic model, it can be assumed that the vibrational amplitude of an atom is constant in all directions. This means that an isotropic
Gaussian probability distribution function is of the form
P( r ) = 1/( √ 2 πσ 2 ) 3 e
-r 2 /(2 σ 2 ) with zero mean and a root-mean-square deviation ∆ r = σ . Using spherical coordinates, the probability of finding an atom inside a sphere of radius r o
centered at the atom's equilibrium position is calculated by
∫
0
2 π
∫
π
0 r
0 ∫
0
1/( √ 2 πσ 2 ) 3 e
-r 2 /(2 σ 2 ) r 2 sin( θ )dr d θ d φ
∫ r
0
0 r 2 e
-r 2 /(2 σ 2 ) dr
= ) e
-r o
2 /(2 σ 2 )
A sphere of radius r o
=1.5382
σ encloses 50 % of the probability density.
4
Once we have an expression for the p.d.f., we can calculate the form of the displacement factor under isotropic conditions (Willis and Pryor, 1975 p. 259 or
Carpenter, 1969 p. 213). As shown in the last section, the displacement factor equals the
Fourier transform of the p.d.f.
ℑ (P( r )) =
∫
V
P j
( r ) e
2 π i r ⋅ s d V
=
2 π π
∫∫∫
∞
0 0 0
1/(2 πσ 2 ) 2/3 e
-r 2 /(2
σ 2 ) e
2 π i r ⋅ s r 2 sin θ dr d θ d φ
= 2 π /(2 πσ 2 ) 3/2
π
∫∫
∞
0 0 e
-r 2 /(2
σ 2 )
e
2 π i r ⋅ s cos θ r 2 sin θ dr d θ
= 2 π /(2 πσ 2 ) 3/2
∞
∫
0 e
-r 2 /(2 σ 2 ) r 2 dr (e 2 π irs -e -2 π irs )/(2 π irs)
= e -2 π
2
σ
2s2
One of the things observed from this calculation is that the Fourier transform of a
Gaussian function is another Gaussian function with zero mean and root-mean-square deviation ∆ s = 1/ σ , allowing us to conclude that ∆ r ⋅ ∆ s = 1.
Under the assumption of isotropic vibration, we obtain as the displacement factor
-
2 π 2 σ 2 s
2 .
ℑ (P( r )) = e
We calculate the structure factor using Equation (1) and minimize the difference between the measured and calculated amplitudes by varying r j
and σ the unit cell.
2 j
for each of the j atoms in
Alternatively, note that s 2 =1/d 2 so, from Bragg's equation 2d sin θ = λ , we get
ℑ (P(
In order to save the trouble of carrying the 8 π the isotropic displacement factor, B, as r )) = e
2
-8 π
2
σ
σ 2
2 sin2
θ / λ
2
term around in the calculations, define
B = 8 π 2 σ 2 .
We now have as our expression for the displacement factor
Bsin2 θ / λ
2
ℑ (P( r )) = e and we minimize the difference between observed and calculated amplitudes by varying r j
and B instead of r j
and σ 2 .
Meaning of σσσσ 2
Let's take a closer look at the meaning of the radial mean-square displacement, <r 2 >.
σ 2 . It has been defined as the mean-square displacement of an atom from its equilibrium position. To understand this, we calculate
<r 2 > ≡
V
∫ P( r ) r 2 dV
5
σ 2
= dV
V
∫
∫
0
2 π π
∫
0
∞
∫
0
-r2/2
σ 2 sin θ dr d θ d φ
∫
∫
∞
0
π
0
∞
∫
0
e -r2/2
σ 2 sin θ dr d θ
σ 2 sin θ dr
The average mean-square displacement of an atom over all space, from its equilibrium position, is <r 2 > = 3 σ 2
If we examine <x 2
.
>, which is the mean-square displacement projected along the x direction we find
<x 2 > ≡
V
∫ P( r ) x 2 dV
∫
V
2
σ 2
= dV
Then, for an isotropic p.d.f. <y 2 > = <z 2 > = <x 2 > = σ 2 .If <x measured along orthogonal axes then
2 >, <y 2 >, and <z 2 > are
<r 2 > = <x 2 > + <y 2 > + <z 2 > = 3 σ 2 .
So, <x 2 > is a projection operator, and the magnitude of vibration in the x direction is actually greater than <x 2 >.
ANISOTROPIC DISPLACEMENT FACTOR
As a second approximation under the harmonic model, it can be assumed that an atom's vibrational displacement varies with direction. The p.d.f. takes the form of an anisotropic Gaussian function:
P( r ) = 1/
(
(2 π ) 3/2 ( σ
1
σ
2
σ
3
)
)
exp
(
-1/2 [ r ] t
C
1 /
0
0
σ 2
1
1 /
0
σ 2
2
0
0
1 /
0
σ 2
3
[ r ]
C
)
. (3)
Equation (3) is written with respect to the Cartesian basis C = { i , j , k } that is coincident with the principal axes of the ellipsoid. Letting [ r ] t t
C
= [s
1 s
2 s
3
C
= [x
1 x
2 x
3
] and [ s ]
], then as discussed earlier, we obtain the displacement factor by taking the Fourier transform of the p.d.f.
ℑ (P( r )) =
∫
V
P j
( r ) e 2 π i r ⋅ s d V
6
1
( 2 π )
= exp
(
-2
( σ
1
σ
2
σ
3
)
π 2 [ s ] t
C
σ
0
0
2
1
∫
V e
− ( x
2
1
/ 2 σ
2
1
+ x
2
2
/ 2 σ
2
2
+ x
2
3
/ 2 σ
2
3
) e 2 π i r ⋅ s d V
0
σ
2
2
0
0
σ
0
2
3
[ s ]
C
)
.
Next we need to transform the exponent to the reciprocal D* basis since it would be awkward to express all the vectors in the crystal with respect to the principal axes of one of the atoms. This is not a necessary step, however it tremendously simplifies the math.
Let [ s ] t
C
. Then
D*
= [h k l] and let M be the matrix such that M[ s ]
D*
= [ s ]
M[a*]
D*
= [a*]
C
giving the first column of M,
M[b*]
M[c*]
D*
= [b*]
D*
= [c*]
C
C
giving the second column of M,
giving the third column of M,
Note that M is not unique but will be different for every translationally non-equivalent atom. Hence
M = [ [a*]
C
[b*]
C
[c*]
C
]
i
k j
⋅
⋅ a a
*
* k i ⋅ b j ⋅ b
⋅ b
*
*
* k i j ⋅
⋅
⋅ c c c
*
*
*
a a a
*
*
* cos(i
= cos(k
∧
∧
∧
cos(i cos(k
∧
∧
∧ a*) a*) a*) a*) a*) a*) b b b cos(i cos(j cos(k
*
*
*
∧
∧ cos(i
∧ cos(j cos(k b*) b*) b*)
∧
∧
∧ b*) b*) b*) c c c
*
*
* cos(i cos(j cos(k
∧
∧
∧ c*) c*) c*)
cos(i cos(j cos(k
∧
∧
∧ c*) c*) c*)
a
0
0
* b
0
0
*
(4) c
0
0
*
defining both C and D. The displacement factor, then, is expressed
ℑ (P( r )) = exp
(
-2 π 2 [ s ] t
C
σ
0
0
2
1
σ
0
2
2
0
0
σ
0
2
3
[ s ]
C
)
= exp
(
-2 π 2 (CD[ s ]
D*
) t
= exp
(
-2 π 2 [ s ] t
D*
D t C t
σ
0
0
2
1
σ
0
0
2
1
0
0
σ
2
2
0
σ
0
2
2
0
σ
2
3
0
σ
2
3
0
0
CD[ s ]
D*
CD[ s ]
D*
)
)
.
7
If U is defined as
U = C t
σ
1
2
0
0
σ
0
0
2
2
0
0
σ
2
3
C, (5) then we obtain the familiar displacement factor expression
ℑ (P( r )) = exp(-2 π 2
= exp ( -2 π 2 (U
11 a*
[ s ] t
D*
D
2 h 2 +U t UD[ s ]
D*
)
22 b* 2 k 2 + U
33 c* 2 l 2 +2U
12 a*b*hk+2U
13 a*c*hl +2U
23 b*c*kl) ) .
The U ij
's are defined as
U
11
U
22
= σ
= σ
U
U
33
12
= σ
= U
1
2
1
2
1
2
21 cos cos cos
=
2
2
2
σ
(
( a * ∧ i ) + σ
( b * ∧ i ) + σ
U
13
= U
31
= σ
1
U
23
= U
32
= σ
1 c
1
2
2
2
* ∧ i ) +
cos(
cos( a a
*
*
σ
∧
∧
2
2
2
2
2
2 i i cos cos cos 2
2
2
) cos(
) cos(
( a * ∧ j ) + σ
( b * ∧ j ) + σ
( c * ∧ j ) + σ b c
*
*
∧
∧ i i )+
)+
σ
σ
2
2
2
2
3
2
3
2
3
2 cos cos cos 2
2
2
( a * ∧ k )
( b * ∧ k )
( c * ∧ k )
cos( a * ∧ j ) cos( b * ∧ j ) + σ
cos( a * ∧ j ) cos( c * ∧ j ) + σ
3
3
2
2
cos( b * ∧ i ) cos( c * ∧ i )+ σ
2
2 cos( b * ∧ j ) cos( c * ∧ j ) + σ
3
2
cos( a * ∧ k ) cos( b * ∧ k )
cos( a * ∧ k ) cos( c * ∧ k )
cos( b * ∧ k ) cos( c * ∧ k )
If we define the matrix β such that β =2 π 2 (D t UD), then
β
β
β
β
11
= 2 π
β
22
= 2 π 2
2 U
11 a*
U
22 b*
2
2
β
33
= 2 π 2 U
33 c* 2
12
= β
21
= 2 π
13
23
= β
= β
31
32
= 2 π
= 2 π
2
2
2
U
U
U
12
13 a*c*
23 a*b* b*c*.
In summary, we have two expressions commonly used for the anisotropic displacement factor:
ℑ (P( r )) = exp(-2 π 2
( -2 π
[ s ] t
D*
D t UD[ s ]
D*
)
=
22 b* 2 k 2 + U
33 c* 2 l 2
+
23 b*c*kl) ) .
( -( β
11 h
33 l 2 + 2 β
12 hk + 2 β
13 hl + 2 β
23 kl) ) .
The β form is popular because of its simplicity.
Symmetry Transformations
If two atoms are related by some symmetry operation, α , then their thermal ellipsoids must also be related by the symmetry operation. Examine this relation for the β matrix form of the displacement parameters. First, note that the translational part of a symmetry operation will not affect the values of the β ij s. Let M
D
( α ) be the matrix representation of the point symmetry operation with respect to the direct basis and
M
D*
( α ) be the representation with respect to the reciprocal basis. Then M vector and let β′ be the transformed β matrix. Then
D
( α )=M
D*
-t ( α )
(Boisen and Gibbs, 1990 p.60). Also, let M
D*
( α )[ s ]
D*
= [ s ′′′′ ]
D*
, where s ′ is the transformed
8
[ s ′ ]
D* t β′ [ s ′ ]
D*
= [ s ]
D* t
(M
D*
β [ s ]
D*
-1 t
( α )[ s ′′′′ ]
D*
) t
(M
D*
-t ( α ) β
β (M
M
D*
D*
-1
-1
( α )[ s ′′′′ ]
D*
)
( α ))[ s ′′′′ ]
D*
.
Since this is true for any vector, s , we can conclude that
β′ = M
D*
-t ( α ) β M
D*
-1 ( α ) = M
D
( α ) β M
D t ( α ).
Example . The O atom located at [.4133, .2672, .1188] with displacement parameters
{ β
11
, β
22
, β
33
, β
12
, β
13
, β
23
} = {.0179, .0130, .0085, .0102, -.0026, -.0041} in α -quartz at
T = 298 K (Kihara, 1990) is transformed to [.1461, -.2672, -.1188] by a 2-fold rotation along a . Find the displacement factors for the transformed O atom. Solution : The rotation matrix is
M
D
( α ) =
1
0
0
− 1
− 1
0 −
0
0
1
,
And so the transformed displacement parameters are found to be {.0105, .0130, .0085,
.0028, -.0015, -.0041}.
Symmetry Constraints
When atoms are known to be in special positions then the displacement factor matrix may reduce to a constrained form. If this is not taken into consideration in a least-squares refinement then singular displacement factor matrices can result. This form can be deduced by setting β′ equal to β in the above expression to obtain the constraint condition
β = M
D
( α ) β M
D t ( α ) where α is the symmetry operation that leaves the atom fixed. In the event that an atom is located on more than one symmetry element, it follows that the constraint condition must be satisfied for each of the symmetry elements. Peterse and
Palm (1965) give a complete list of constrained forms.
Example . The Si atom at [.4697, 0, 0] is located on a 2-fold rotation axis that is parallel to a in α -quartz (Kihara, 1990) at 298 K. Thus its displacement parameters are constrained such that
β
11
β
12
β
13
β
12
β
22
β
23
β
β
13
23
β
33
=
1
0
0
−
−
0
1
1
−
0
0
1
β
11
β
12
β
13
β
β
β
12
22
23
β
β
13
23
β
33
−
1
0
1 −
β
11
−
β
2
23
β
−
12
β
β
+
12
13
β
22
β
22
β
−
23
β
12
β
23
β
β
−
23
33
β
13
0
0
1
.
−
0
0
1
Solving, we obtain the constraints that 2 β
13
= β
23
, and 2 β
12
= β
22
, and one of the principal axes of the ellipsoid must lie parallel to the a -axis.
Mean-Square Displacements Along Vectors
To obtain an expression for the mean-square displacement, < µ 2 v
>, of an atom as projected along some specified vector, v , assume a Cartesian basis, C = { i j k }, coincident with the principal axes of the ellipsoid (Nelmes, 1969). Let [ v ]
C t = [v
1
v
2
v
3
] and the dummy vector [ r ]
C t =[x
1
x
2
x
3
] then
9
< µ 2 v
> =
V
∫
( v / || v || ⋅ r ) 2 P( r ) d V
( 2 π )
1
( σ
1
σ
2
σ
3
) || v || 2
∫
V
( v ⋅⋅⋅⋅ r ) 2 e
− ( x
2
1
/ 2 σ
2
1
+ x
2
2
/ 2 σ
2
2
+ x
2
3
/ 2 σ
2
3
)
|| v
1
||
C t
σ
1
0
2
0
[ v
[
] t v
D
]
* t
D *
D t
G
UD
* [
[ v ] v ]
D *
D *
[ v ] t G
[ v t
]
D
D t UDG
G [ v ]
D
[ v ]
D
0
σ 2
2
0
0
0
σ 2
3
[ v ]
C
=
[
2 v
π
] t
2
D
[
G v t β
] t
D
G [
G [ v ]
D v ]
D d V
Example . Compute the mean-square displacement amplitude of the O atom along the
SiO vector in α quartz at 298 K (Kihara, 1990). Solution : If O is at [.4133, .2672, .1188] and Si is at [.4697, 0, 0], then [ v ]
D t = [.0564, -.2672, -.1188]. The displacement parameters for O are { β
11
, β
22
, β
33
, β
12
, β
13
, β
23
} = {.0179, .0130, .0085, .0102, -.0026,
-.0041}. The cell parameters are a = 4.9137 Å, c = 5.4047 Å, so the metrical matrix is
G =
−
24 .
14445
12 .
07222
0
− 12 .
07222
24 .
14445
0
0
0
29 .
21078
.
Then < µ 2 v
> = 0.006935 Å 2 .
Principal Axes
It is of interest to determine the length and orientation of the principal axes of a thermal ellipsoid. Since the probability ellipsoid has quadratic form ½ x t Λ x where Λ may be diagonalized to
Λ =
1 /
0
0
σ 2
1
1 /
0
σ 2
2
0 1 /
0
0
σ 2
3
, then the lengths of the principal axes must be σ
1
, σ
2
, and σ
3
(Franklin, 1968 p.94). We can obtain these three optimum values of σ by considering Equation (6)
< µ 2 v
> =
[ v ] t
D
2 π 2 [
G v t β
] t
D
G [
G [ v ] v ]
D
D .
The strategy to obtain the solution is to recognize that the principal axes are coincident with directions of critical points in the values of < µ 2 v
>. That is, < µ 2 v
> has its maximum and minimum values along the principal axes. Note that this discussion will use the β form of the displacement parameters, however the method applies equally well to the U
10
form (see Waser, 1955 or Busing and Levy, 1958). Take the derivative of < µ respect to the vector v , and set it to zero,
2 v
> with d < d
µ v
2
V
>
=
( 2 G t β G [ v ]
D
)( 2 π 2 [ v ] t
D
(
G [ v
2 π 2 [
]
D
) v ] t
D
−
G
(
[
4 π v ]
2
D
G [
) 2 v ]
D
)([ v ] t
D
G t β G [ and so
(2G t β G[ v ]
D
)(2 π 2 [ v ]
(2G t
D t
β
G[ v ]
D
) = (4 π 2
G[ v ]
D
) = (4 π 2
G[ v ]
D
G[ v ]
D
)([ v ]
D t
)
([
( 2
G v ] t
π 2
D
[ t
G v
β G [ v ]
] t t
β
D
G
G [
[ v v
D
)
]
D
]
D
)
) v ]
D
)
= 0,
β G[ v ]
D
= λ [ v ]
D, where λ = 2 π 2 σ 2 .
The root-mean square displacements parallel to the principal axes of the ellipsoid are the solutions to the equation σ = √ ( λ /2 π 2 ) for the three eigenvalues of β G. The eigenvectors are parallel to the principal axes and are expressed in direct space.
Example . Determine the lengths and directions of the principal axes of the Si atom in
α quartz at 298 K (Kihara, 1990). The cell parameters and the metrical matrix were given in the last example, and { β
-.00015, -.0003}, thus β G is
11
, β
22
, β
33
, β
12
, β
13
, β
23
} = {.0080, .0061, .0045, .00305,
β G =
=
The eigenvalues are
−
.
0080
.
00305
.
00015
.
156335
.
000000
.
000000
.
00305
.
0061
− .
0003
− .
022937
.
110461
− .
005433
−
−
.
00015
.
0003
.
0045
−
−
.
.
004382
.
008763
131449
−
24 .
14445
12 .
07222
0
.
− 12 .
07222
24 .
14445
0
0
0
29 .
21078
.10838, .13337, .15635, and the unit length eigenvectors in direct space coordinates are
.
.
1126
2251
.
0531
,
−
−
.
.
0337
.
0674
1773
,
.
2035
0
0
.
The root mean square lengths of the axes are found from λ = 2 π 2 σ 2 to be
.0741 Å, .0822 Å, .0890 Å.
Note that the principal axes define an ellipse that is close to spherical. This is common for Si atoms in tetrahedral coordination under the harmonic approximation. It will be shown later that the displacement factors of the Si atoms in quartz primarily result from translational effects under the rigid-body model for vibration of the SiO
4
group.
To further define the eigenvectors, many authors publish their orientations with respect to the direct crystal basis. These angles are obtained by computing the dot product of the eigenvectors with the basis vectors.
11
Example . For the ellipsoid in the last example we obtain
∠ ( v
∠ ( v
∠ ( v
1
2
3
∧ a ) = 90 °
∧ a ) = 90 °
∧ a ) = 0 °
∠ ( v
∠ ( v
∠ ( v
1
2
3
∧ b ) = 33.9
° ∠ ( v
∧ b ) = 104.4
° ∠ ( v
1
∧ b ) = 120 ° ∠ ( v
3
2
∧ c ) = 73.3
°
∧ c ) = 16.7
°
∧ c ) = 90 ° .
Obtaining Ellipsoid Parameters From Principal Axes Information
To calculate the U ij s or β ij s from the rms amplitudes and orientations of the principal axes can be extremely useful. For instance, there was a period of time during which the
American Mineralogist did not publish many displacement parameters but instead published the principal axis information such as that worked out in the example of the last section. However, this information is of limited use since the original displacement parameters are necessary to do calculations. Fortunately, it is possible to retrieve the displacement parameters from the principal axes information. Furthermore, if tables of both displacement parameters and the rms and orientations of the principal axes are given, then typographical errors in the displacement parameters can be corrected by calculating them back from the principal axes information.
The procedure is straightforward. In Equation (5) U has been defined as
U = C t
σ 2
1
0
0
0
σ 2
2
0
0
0
σ 2
3
C,
Hence, we need only to determine the matrix C, defined earlier as
C =
cos(i cos(j
cos(k
∧
∧
∧ a*) a*) a*) cos(i cos(j
∧
∧ b*) b*) cos(k ∧ b*) cos(i cos(j cos(k
∧
∧
∧ c*) c*) c*)
, where i , j , and k are the normalized principal axes vectors. Suppose we write the principal axes information in matrix format as
∠
∠
∠
(i
(j
(k
∧
∧
∧ a) a) a)
∠ (i ∧
∠ (j ∧ b) b)
∠ (k ∧ b)
∠ (i
∠ (j
∠ (k
∧
∧
∧ c) c) c)
Then, if we take the cosine of each term and multiply by the cell parameters, we obtain
acos(i acos(j
acos(k
∧
∧
∧ a) a) a) bcos(i ∧ b) bcos(j ∧ b) bcos(k ∧ b)
If we transform to reciprocal space then and we find that
k j i
⋅
⋅ a
⋅ a a i ⋅ b j ⋅ b k ⋅ b ccos(i ccos(j ccos(k
∧
∧
∧ c) c) c)
k i j ⋅
⋅
⋅ c c c
G* =
k j i
⋅
⋅ a
⋅ a a
*
*
*
= i ⋅ b * j ⋅ b * k ⋅ b *
k j i
⋅
⋅ a
⋅ a a i ⋅ b j ⋅ b k ⋅ b i k j ⋅
⋅
⋅ c c c
*
*
*
k i j ⋅
⋅
⋅ c c c
12
C =
k j i
⋅
⋅
⋅ a a a i ⋅ b j ⋅ b k ⋅ b i k j ⋅
⋅
⋅ c c c
G*
1/a
0
0
* 0
1/b *
0
0
0
1/c *
Example . The example in the previous section can be worked backwards to obtain the initial displacement parameters.
Cross-Sections of the Ellipsoids
Calculations to obtain thermal corrected bond lengths (Busing and Levy, 1964a) need the mean-square displacement of an atom in the plane perpendicular to a given bond vector. Given the vector between any two bonded atoms, v , we can calculate the amplitude vibration along the bond using Equation (6),
< µ 2 v
> =
[v] t
D
G t
D t
UDG[v]
D
[v] t
D
G[v]
D
=
[v]
2 π t
D
2
G
[v] t
β G[v] t
D
G[v]
D
D
.
The mean square displacement for the atom, over all space, is the sum of the displacements along the principal axes of the ellipse and hence can be expressed
<r 2 > = σ
1
2 + σ
2
2 + σ
3
2 = trace( β G)/2 π 2 .
If we imagine a change of basis such that the bond vector coincides with one of the new basis axes, then by invariance of the trace under similarity transformations, the isotropic mean-square displacement of the atom in a plane perpendicular to the bond vector is
<r ′ 2 > = trace( β G)/2 π 2 - < µ 2 v
>.
This is the equation used in the ORFFE program (Busing et al, 1964) to calculate the parameters for the riding correction to bond lengths. However, this method obscures the anisotropic information about the shape and orientation of the cross-section. To obtain this additional information let v be any vector which is perpendicular to the desired crosssection, where [ v ]
D t = [v
1
v
2
v
3
] with respect to the direct basis and [ v ]
[v
1
* v
2
* v
3
*] with respect to the reciprocal basis. If x, ([ x ]
D t in the cross-section, then the equation
D* t = (G[ v ]
D
) t =
= = [x
1
x
2
x
3
] is any vector x ⋅⋅⋅⋅ v = [ x ]
D t G[ v ]
D
= [ x ]
D t [ v ]
D*
= x
1 v
1
* + x
2 v
2
* + x
3 v
3
* = 0 must be satisfied. Suppose q and r are any two non-collinear vectors satisfying this equation. Then we can obtain the elliptic cross-section by transforming the ellipsoid to the constrained plane using a transformation matrix, T, with [ q ]
D
and [ r ]
D
as its columns,
T =
[
[ q ]
D
[ r ]
D
]
.
T will transform a vector from the plane, written with respect to the basis D ′ = { q , r }, to our three-dimensional direct space. If v
3
* is not zero then q and r can be chosen as follows. A solution for x
3
can be written in terms of x
1
and x
2
as x
3
= − v v
1
3
*
* x
1
− v v
2
3
*
*
x
2
.
Choosing q
1 equation
= 1, q
2
= 0 for q and r
1
= 0, r
2
= 1 for r we obtain a transformation
13
T[ x ]
D ′
=
( − v
1
1
0
* / v
3
*) ( − v
2
0
1
* / v
3
*)
x x
1
2
=
x x x
1
2
3
= [ x ]
D
.
Any choice of non-collinear q and r is theoretically satisfactory, however for computational purposes, in which numerical instability may be a problem, it may be best to choose them to be orthonormal.
After the transformation matrix has been chosen we can then obtain an expression for displacements in the plane,
<u 2 > =
[ x
2 π
] t
2
D
[
G x t
]
β G [ x t
D
G [ x
]
D
]
D
2
( T [
π 2 ( x
T
]
[
D ′ x
)
] t G
D ′
) t t
β
G
GT
( T
[
[ x x
]
]
D ′
D ′
)
[ x
2 π
] t
2
D ′
[
T x ] t G t
D ′ t
T
β GT
GT
[
[ x x
]
]
D ′
D ′
To obtain the principal axes of this ellipse and the associated eigenvalues we solve the generalized eigenvalue problem (Franklin, 1968),
T t G t β GT[ x ]
D ′
= λ T t GT [ x ]
D ′
.
[ x i
]
D ′ t = [x ′
1i
x ′
2i
] are the two eigenvectors and λ i
= 2 π 2 σ′ i
2 are the two eigenvalues. It follows that in direct space the principal axes of the ellipse perpendicular to the vector v are T[ x i
]
D ′
. In addition, it is found that
<r ′ 2 > = trace( β G)/2 π 2 − < µ 2 v
> = σ′
1
2 + σ′
2
2 .
Example . Again, using the previous example for Si, we find that the mean-square displacement amplitude along the SiO bond is .006354 Å plane perpendicular to the SiO bond are .006647 Å 2
2 , while the principal axes in the
and .007174 Å 2 .
Surfaces
If Equation (6) was used to calculate the root-mean squared displacements, < µ for all possible vectors emanating from the center of the ellipsoid, v
2 > 1/2 ,
< µ 2 v
> =
[v] t
D
G t
D t
UDG[v]
D
[v] t
D
G[v]
D
=
[v]
2 π t
D
2
G
[v] t
β G[v] t
D
G[v]
D
D
, then the resulting surface would be a peanut shaped quartic as displayed in Figure 4.
Hummel et al (1990a,b) have written a software program called PEANUT to facilitate rendering thermal parameters in this fashion. Jürg Hauser at Universität Bern currently maintains the software. The information obtained from rendering quartic surfaces is the same as that obtained from ellipsoidal surfaces, except that the quartic surface remains closed when the harmonic parameters are non-positive definite. Otherwise, manipulation of Equation (3) can provide an ellipsoidal surface of constant probability
14
[ v ]
C t
1 /
0
0
σ 2
1
1 /
0
σ 2
2
0
0
1 /
0
σ 2
3
[ v ]
C
= K 2 (7) expressed with respect to the Cartesian basis C = { i , j , k } coincident with the principal axes of the ellipsoid. When K = 1.5382 then the ellipsoid contains 50% of the probability density. Now, transform this equation into the crystal basis,
Therefore, we find that
β = 2 π 2 (D t UD) = 2 π 2 D t C t
σ 2
1
0
0
1 /
0
0
σ 2
1
1 /
0
0
σ 2
2
0
σ 2
2
0
0
0
σ 2
3
1 /
0
0
σ 2
3
=2 π 2 CD β -1 D t C t .
CD.
Substitution into Equation (7) gives
D t C t [ v ]
C
= K 2 .
But CD[ v ]
D*
= [ v ]
C
, so
D t
Figure 4.
An image of the displacement quartics for a SiO
4 quartz, SiO
2
group in α -
, at 498 K (Kihara, 1990). The image was kindly rendered by
Jürg Hauser with the PEANUT software package.
15
Isotropic Equivalent to the Anisotropic Displacement Factor
In some instances, it is of use to know the isotropic displacement factor that would be equivalent to the anisotropic one. The equivalent isotropic factor is not usually the one that would be obtained from a least-squares refinement, but it can be considered a good estimate. It is a parameter that is commonly published because it takes less space than the anisotropic parameters, and it offers a convenient way to assess the nature and quality of data. In practice, assume that the isotropic equivalent mean-square displacement is equal to the average of the mean-square displacements along the principal axis of the anisotropic thermal ellipsoid. Recalling that λ i
= 2 π 2 σ i
2 are the eigenvalues of β G, and that the trace of this matrix is invariant under a similarity transformation, then the average eigenvalue of β G is 1/3 trace ( β G). Hence the average mean square displacement is
< σ 2 > =
3 ⋅
1
2 π 2 tr( β G).
The equivalent isotropic displacement factor is then
B eq
= 8 π 2 < σ 2 > (8)
= 4/3 tr( β G) = 4/3
3 ∑ 3 ∑
β ij
G ij
= 4/3 tr(2 π 2 D t i = 1
UDG) = 8 π 2 j = 1
/3 tr(D t UDG).
Please note that there has been confusion propagated in many papers about how to calculate this factor when the anisotropic factors have been presented in the U form. The expression B bases. eq
= 8 π 2 /3 tr(U) is commonly used, but this is only valid for orthogonal
Example . Calculate the equivalent isotropic displacement factor for Si in quartz at
298 K (Kihara, 1990). Using the β G matrix from the example in the section on principal axes, we obtain
B eq
= 4/3 tr( β G) = 4/3 (0.156335 + 0.110461 + 0.131449) = 0.531.
As a check, we can take the average from the root mean square lengths of the principal axes (these are calculated in the section on Principal Axes).
B eq
= 8 π 2 < σ 2 > = 8 π 2 (0.0741
2 + 0.0822
2 + 0.0890
2 )/3 = 0.531.
Anisotropic Equivalent to the Isotropic Displacement Factor
Sometimes we may only know the isotropic displacement factor and may want an estimate of the anisotropic matrix. For instance, in a least-squares refinement procedure one often refines for the isotropic case first and then expands to the anisotropic. Estimates for starting parameters may be needed.
Define β eq
as our estimated anisotropic matrix. Then, by modifying Equation (8),
¾ B = tr( β eq
G).
Converting to 3-dimensional space,
β eq
G = B/4 I,
16
where I is the 3x3 identity matrix, in order to constrain the ellipse to a spherical shape.
Hence
β eq
= B/4 G*.
Example . For the Si atom in α quartz at 298 K (Kihara, 1990)
β eq
= B/4 G*
=
4
0
0
.
05522
.
02761
0
0 .
02761
0 .
05522
0
0
0
0 .
03423
=
0
0
.
00733
.
00367
0
0 .
00367
0 .
00733
0
0
0
0 .
00454
.
RIGID BODY MOTION
The earlier part of this chapter examined the harmonic motion of independent atoms.
However, if the bonds in a group of atoms are strong enough, say for instance in a SiO
4 group, then the entire group may undergo harmonic oscillation of a type called rigid body motion. A group of atoms oscillating with rigid body motion undergo correlated translational and librational motion as a group. The motion of each individual atom is determined entirely by the motion of the group. The translational motion of the group is the same motion as assumed for the independent atom and can be described in the same fashion as laid out in the previous section of this chapter. The librational motion is angular oscillation about a line that usually passes near the center of mass of the group.
Furthermore, the translational and librational motions may be coupled in such a way that the net effect is a sort of screw motion. A strong screw component is typical of the motion of CO
3
groups in carbonates (Finger, 1975; Markgraf and Reeder, 1985; Reeder and Markgraf, 1986). Together these three are known as the TLS model of rigid body motion (Schomaker and Trueblood, 1968). At this point in time, no one has successfully coded a computer program that directly refines the rigid body parameters for any crystal.
Several attempts have been made, but they do not work in the general case (cf. Finger and
Prince, 1975; Finger, 1975; Markgraf and Reeder, 1985; Downs et al, 1996).
Consequently, it is standard practice to compute the rigid body parameters from the anisotropic parameters (cf. Ghose et al, 1986; Downs et al, 1990; Stuckenschmidt et al,
1993; Bartelmehs et al, 1995; Jacobsen et al, 1999).
The study of the motion of rigid bodies constitutes an important part of physics and much work has been done in this area (cf. Landau and Lifshitz, 1988). The first important paper considering the thermal vibrations of a group of atoms was Cruickshank (1956). He proposed a model that included translation and libration about the center of mass of a molecule. Schomaker and Trueblood (1968) showed that Cruickshank’s model was but a special case, valid only if the origin of the rigid body is at an inversion center, of the more general situation where the libration axes may be non-intersecting and, in fact, the motion may best be described as spiral or screw motions. Around the same time period,
Brenner (1967) tackled the problem with application to the Brownian motion of rigid particles. Both of these papers treat the mathematics with a tensor of dyadic notation, and
Johnson (1969) is recommended for an alternate review of the subject using matrix notation.
Rigid body criteria
17
Suppose there exists a rigid group of N atoms. The term rigid means that the separations between all atoms in the group remains constant, regardless of the overall motion of the group. This implies that the atoms in a rigid group vibrate in tandem, and therefore the thermal parameters of the N atoms represent the same motion. It is questionable whether such a group truly exists in nature, however, we find that many groups of atoms satisfy the definition of rigidity within the accuracy of crystal structure refinements (Finger, 1975; Markgraf and Reeder, 1985; Reeder and Markgraf, 1986;
Ghose et al, 1986; Downs et al, 1990; Stuckenschmidt et al, 1993; Bartelmehs et al, 1995;
Jacobsen et al, 1999).
If a group of atoms is rigid then all the atoms in the group vibrate in tandem. This implies that the mean-square displacement amplitudes exhibited by any pair of atoms in the group should be equal along their interatomic vector (Dunitz et al, 1988). Define the difference displacement parameter, ∆
AB atoms, A and B, as
, evaluated along the vector, v , between two
∆
AB
= < µ 2 v
D* t
>
B
- < µ 2 v
>
A
2 π
Then
4
groups a suitable tolerance for the SiO bonds has been proposed by Downs et al (1990) as -0.00125 Å
Å 2
∆
AB
should be zero, within some tolerance. For SiO
, and between the OO atoms as ∆
OO
= |< µ 2 v assumed that the group behaves as a rigid body.
>
O1
− < µ 2 v
>
O2
| ≤ 0.003 Å
2
2
≤ ∆
SiO
≤ 0.002
(Bartelmehs et al, 1995). If the ∆
AB
values for a group of atoms lie within such tolerances, then it can be
Example . The ∆
AB
values for the SiO
4
tetrahedra in quartz as a function of temperature (Kihara 1990) are computed and given in Table 1. The values for α -quartz represent averages over the non-equivalent vectors. The results tabulated in Table 1 demonstrate that the SiO
4
group in both α and β -quartz can be considered to vibrate as if a rigid body over the temperature range 298 K ≤ T ≤ 1078 K.
Table 1. The ∆
AB
values for the SiO temperature (Kihara 1990).
4
tetrahedra in quartz as a function of
T (K) ∆
SiO
(Å 2 ) ∆
OO
(Å 2 )
α -quartz
0.00062
0.00065
0.00087
0.00132
0.00148
838 0.00110
β -quartz
0.00000
18
0.00000
0.00000
1012 0.00167 0.00000
1078 0.00069 0.00000
Libration of a body
If a rotation of θ takes place about the unit vector l = l
1 i + l
2 j + l
3 k , expressed with respect to the Cartesian basis, C = { i , j , k }, then the general Cartesian rotation matrix,
M
C
( θ ), describing the transformation of a vector v to v ′ , M
C
( θ ) v = v ′ , can be expressed as
M
C
( θ ) =
l l
1
1 l l l
1
2
2
3
(
( 1
1
( 1
−
−
− cos cos cos
θ
θ )
θ )
) +
+
− l cos l
2
3
θ sin sin
θ
θ l l
1 l
2
( 1 l 2
2
( 1
−
− cos cos
θ )
θ )
−
+ l
3 sin cos θ
θ
2 l
3
( 1 − cos θ ) + l
1 sin θ l
1 l
3
( 1 − l
2 l
3 l
3
2
( 1
( 1
−
− cos cos
θ
θ
)
) cos θ )
+
−
+ l l
2
1 sin sin cos θ
θ
θ
.
This expression can be rewritten as
M
C
( θ ) = I + sin θ L + (1-cos θ ) L 2 , where
L =
−
0 l
3 l
2
− l
3
0 l
1
− l
2
0 l
1
and L 2 =
− l
2
2 l
1 l
2 l
1 l
3
− l
3
2
− l
1 l l
1
2
2
− l
3
2 l
2 l
3 l
1 l
3
− l
2 l
1
2 l
3
− l
2
2
.
Expand sin θ and cos θ in a Taylor series about θ = 0, and note that L = − L and L 2 = – L 4 = L 6 = – L 8 …, then M
C
( θ ) can be rewritten as
3 = L 5 = − L 7 …,
M
C
( θ ) = I + θ L +
θ 2
2
L 2 +
θ 3
6
θ 4
L 3
24
L 4 + ….
If the axial vector λλλλ (Johnson, 1970) is defined as λλλλ = θ l , then
M
C
( θ ) = I +K +
1
2 !
K 2 +
1
3 !
K 3 +
1
4 !
K 4 + …, where
K =
−
λ
0
λ
3
2
−
λ
0
λ
1
3
−
λ
0
2
λ
1
.
We can define the displacement of a particle, v = r ′ - r , as v = K r +
1
2
K 2 r +
1
6
K 3 r +
1
24
K 4 r + …,
19
1
2
λλλλ× ( λλλλ× r ) +
1
6
λλλλ× ( λλλλ× ( λλλλ× r )) +
1
24
λλλλ× ( λλλλ× ( λλλλ× ( λλλλ× r ))) + ….
The quadratic term is λ× r, which gives an error of about 2% for θ = 20 ° . With a cubic truncation of the series, the error is only about 0.01%.
The quadratic approximation for rigid body motion
In the quadratic approximation, the displacement, u , of a rigid body can be separated into two parts as u = t + λλλλ× r . (9)
The vector, t , represents a translational component of motion. Every part of a rigid body has the same translational component. The term λλλλ× r represents the librational component of motion of the part of the body that is located at the end point of the vector r , about an axis of rotation λλλλ . The vector λλλλ represents the direction of the rotational axis and the magnitude of λλλλ represents the libration angle. Both r and λλλλ originate from the same arbitrary origin.
Given any position, r , in the rigid body, the displacement at that position can be described with a 6 component vector, c t = [t
1
t
2
t be rewritten, with respect to a Cartesian basis, as
3
λ
1
λ
2
λ
3
] = [t M
λ ]. Equation (9) can then
u u
2
u
1
3
=
1
0
0
0
1
0
0
0
1
− r
0
2 r
3
− r
0
3 r
1
− r
1
0 r
2
t t
λ
λ
1
2 t
λ
3
1
2
3
t
λ
.
The atomic displacement parameters, U, for a given atom located at position r can be obtained by taking the time average of the outer product of the displacement vector,
< u ∗ u > = < uu t >, for that atom,
U = < u ∗ u > =
<
<
< u u u
1
1
1 u u u
1
2
3
>
>
>
<
<
< u u u
1
2
2 u u u
2
2
3
>
>
>
<
<
< u u u
1
2 u u
3 u
3
3
3
>
>
>
.
Note that the values of the components of this matrix are basis dependent. From
Equation (9) we obtain
U = < u ∗ u > = <( t + λλλλ× r ) ∗ ( t + λλλλ× r )>
=
=
≡
< t t ∗
∗ t t > + <
> + < t t
T + AS + S
∗
∗ t
( λλλλ×
A
A t
λλλλ r )> + <(
> + <A
+ A L A t
λλλλ×
λλλλ∗ t r ) ∗ t > + <(
> + <A λλλλ∗ A
λλλλ×
λλλλ r
>
) ∗ ( λλλλ× r )>
(10)
20
Method for obtaining the TLS rigid body parameters
The method for calculating the T, L and S matrices that describe the motion of a rigid molecule will now be shown. Using Equation (10), we set up 6 equations, one for each of the independent U ij s, and then apply simple linear regression methods to obtain the T ij s,
L ij s, and S ij s.
U
11
= T
11
+ r
3
2 L
22
+ r
2
2
U
22
U
33
U
U
U
12
13
= T
22
= T
33
= T
23
= T
12
= T
13
23
+ r
+ r
– r
– r
1 r
3
– r
3
2
2
2
1
2 r
2
L
11
L
11
L
33
+ r
– r
L
22
+ r
2 r
3
L
11
+ r
1
2
1
2
2
3
+ r
1 r
L
33
– 2r
2 r
3
L
23
+ 2r
3
S
21
– 2r
2
S
31
L
L
33
– 2r
22
– 2r
+ r
1
1 r
3
L
1 r
2
2
L
13
– 2r
12
+ 2r
3
2
S
S
12
13
+ 2r
1
– 2r
S
32
1
S
23
L
12
+ r
2 r
3
L
13
+ r
1 r
3
L
23
+ r
3
(S
22
– S
11
) + r
1
S
31
– r
2
S
32
3 r
3
L
12
L
12
– r
2
2 r
L
13
+ r
1
L
13
– r r
1
2
2
L
23
L
23
+ r
2
+ r
1
(S
11
(S
33
– S
33
– S
22
) – r
1
) + r
2
S
21
S
12
+ r
3
S
23
– r
3
S
13
Observe that the parameters S
11
, S
22
, and S
33
only appear in the combinations (S
22
–
S
11
), (S
11
– S
33
) and (S
33
– S
22
) in the U
12
, U
13
, and U
23
terms. This imposes a constraint on the values of S
11
, S
22
and S
33
which can be seen by observing that if all parameters were zero, except for these, then the following equation would need to be solved:
U
12
U
U
13
23
=
− r
2
0 r
3
− r
0
3 r
1
−
0 r
1 r
2
S
11
S
S
22
33
.
The determinant of the matrix is zero, indicating that the S
11
, S
22
, and S
33
terms are not linearly independent. By convention, the constraint is imposed that S
11
+ S
22
+ S
33
=
0. Hence eliminating the S
33
term, we solve the 20 parameter problem
U
11
= T
11
+ r
3
U
U
U
U
U
22
33
12
13
23
= T
22
= T
= T
33
12
+ r
+ r
– r
= T
13
– r
1
= T
23
– r
2
2
3
2
2
2
1 r r
3
2
L
22
+ r
2
2
L
11
L
11
L
L
33
22 r
3
L
11
+ r
+ r
– r
+ r
1
2
1
2
2
3
+ r
1
2 r
L
33
– 2r
2 r
3
L
23
+ 2r
3
S
21
– 2r
2
S
31
L
L r
3
3
33
22
– 2r
– 2r
L
12
1
1 r
L
12
+ r
1 r
2
3
L
2
L
13
– 2r
12
+ 2r
3
2
S
S
12
+ 2r
1
13
– 2r
1
S
32
S
23
L
12
+ r
2 r
3
L
13
+ r
1 r
3
L
23
– r
3
S
11
+ r
3
S
22
+ r
1
S
31
– r
2
S
32
– r
2
2 r
L
13
+ r
L
13
– r
1 r
1
2
2
L
23
L
23
+ 2r
2
– r
1
S
S
11
11
– r
1
S
+ r
2
S
12
21
+ r
2
S
22
– r
3
S
13
(11)
+ r
3
S
23
– 2r
1
S
22
There will be one set of each of these equations for each atom in the rigid molecule, so that in the most general case, with 20 unknowns and no symmetry constraints, we need at least 4 atoms in the molecule to solve the problem.
Choice of Origin
The choice of origin is arbitrary, but affects the values of the T and S parameters. No matter where the origin is chosen, λλλλ and r must originate from that point. Hence the extent of the displacement due to libration can vary, depending upon the magnitude of λ
× r. Since the final solution is independent of origin, the librational displacement can be changed by altering the correlation between translation and libration, and by altering the translation parameters. There exists a unique choice of origin that minimizes the trace of
T. That is, the isotropic magnitude of translation is minimized. This origin is called the center of diffusion by Brenner (1967), and the center of reaction by Johnson (1969).
From Equation (11),
Tr(T) = T
11
+ T
22
+ T
33
U
11
– (r
3
2 L
22
+ r
2
2 L
33
– 2r
2 r
3
L
23
+ 2r
3
S
21
– 2r
2
S
31
)
21
+ U
22
– (r
+ U
33
– (r
2
3
2
2
L
L
11
11
+ r
+ r
1
2
2
1
L
33
– 2r
1 r
3
L
13
– 2r
3
S
12
+ 2r
1
S
32
)
L
22
– 2r
1 r
2
L
12
+ 2r
2
S
13
– 2r
1
S
23
).
Then Tr(T) is minimized with respect to a choice of origin if
∂ Tr ( T
∂ r
1
∂ Tr ( T
∂ r
2
∂ Tr ( T
∂ r
3
)
)
)
= –r
1
(L
22
+ L
33
) + r
2
L
12
+ r
3
L
13
– S
32
+ S
23
= 0,
= r
1
L
12
– r
2
(L
11
+ L
33
) + r
3
L
23
– S
13
+ S
31
= 0,
= r
1
L
13
+ r
2
L
23
– r
3
(L
11
+ L
22
) – S
21
+ S
12
= 0.
The center of reaction must then satisfy the equation
r r
2 r
3
1
=
( L
22
−
−
+
L
L
12
13
L
33
)
( L
11
− L
12
+ L
33
)
− L
23
− L
13
( L
11
− L
23
+ L
22
)
− 1
S
23
S
S
31
12
−
−
− S
32
S
13
S
21
.
We also see that a shift to this origin causes the S matrix to be symmetrized since r
1
= r
2
= r
3
= 0 in the new setting, insuring that S
23
– S
32
= S
31
– S
13
= S
12
– S
21
= 0.
Example . The atomic parameters for both α and β -quartz were used to refine TLS parameters as a function of temperature. The procedure is to obtain the coordinates and displacement parameters for the 5 atoms in a SiO
4
group. These are transformed to
Cartesian coordinates, with the origin chosen at the Si atom. The Cartesian system is chosen such that the z -axis is parallel to c , and x -axis is in the ac plane. TLS parameters are refined in a linear least-square process that minimizes the differences between the observed U ij s and the ones calculated with Equation (11). With this solution, the center of reaction can be computed, and the problem can be solved again at the new choice of origin. The resulting TLS parameters for β -quartz at 848 K are
(1,1) (2,2) (3,3)
0.02020(5)
L 0.02973(6) with the off-diagonal terms all equal to 0. The observed and calculated displacement parameters are
U(1,1) U(2,2) U(3,3) U(1,2) U(1,3) U(2,3)
Si
Si
O obs.
O calc.
0.0521
0.0521
0.0576
0.0576
0.0388
0.0388
0.02605
0.02608
0
0
0
0
0
0
-.0269
-.0269
22
The parameters for the other 3 O atoms can be obtained by applying the appropriate symmetry transformation. Examination of these results demonstrates the successful fitting of the thermal motion of β -quartz to a rigid body model at 848 K.
Interpretation of Rigid Body Parameters
The translational component of the TLS model is straightforward to interpret. It represents the translational motion of the entire group of atoms, just as the anisotropic displacement parameters discussed in the previous section of this chapter represent the motion of independent atoms. Therefore, the interpretation can be carried out in the same manner. Since the translational parameters are computed in Cartesian coordinates, the math is a little easier.
Example . Compute the isotropic equivalent translational motion for the SiO
β quartz at 848 K. The isotropic equivalent is determined from the trace of T as
4
group in
B eq
= 8 π 2 < σ 2 > = 8 π 2 (0.02868 + 0.02020 + 0.02001)/3 = 1.81 Å 2 .
The observed B eq
(Si) at 848 K for β -quartz is 1.80 Å 2 (Kihara, 1990), and is statistically equivalent to the B eq
of the translational motion for the entire SiO
4
group. In general, this result is typical for SiO
4
groups. The motion of the Si atom is entirely translational, and the T matrix is more or less equal to the displacement parameters of the Si atom
(Bartelmehs et al, 1995).
The interpretation of the librational component of motion is straightforward, though not as easy as for the translational part (Bartelmehs, 1993). If the eigenvalues of L are positive, as they should be, then the L matrix represents an ellipsoid. Define the normalized eigenvectors of L as libration (in radians) is θ = ( λ l
1 respectively. Then the libration vector, λλλλ , is λλλλ = λ
1 l
1
+ λ
2 l
2
+ λ
3 l
3
. The magnitude of
2
1
, l
2,
+ λ 2 and l
2
+ λ 2
3
3
, associated with eigenvalues λ
) 1/2 = trace(L) 1/2 matrix U with columns constructed from the eigenvectors
2
1
, λ 2
2
, and λ 2
3
,
. Define the transformation
U = [
[ l
1
]
C
[ l
2
]
C
[ l
3
]
C
] , where C is the Cartesian basis chosen to solve the TLS refinement. Then U represents a l linear transformation from the Cartesian space defined by the eigenvectors of L ( L = { l
1
,
2, l
3
}) to the Cartesian space defined by C, such that U[ v ]
L
= [ v ]
C
.
Example . Determine the magnitude of libration and the libration vector for the SiO
4 group in β quartz at 848 K. The L matrix was determined in the last example. Solution :
Since L is already a diagonalized matrix then the eigenvectors are already determined.
Therefore the magnitude of libration, θ , is
θ = (.02973 + .01635 + .01107) 1/2 = .239 rad = 13.7
° .
The axis of libration is found, first by constructing U. In this case, since L is a diagonalized matrix, then the eigenvectors are i , j , k and U is the identity. In general, however, you must solve the eigenvalue problem. If l is the normalized vector parallel to
λλλλ , then
[l ]
C
= U [ l ]
L
23
1
0
0
1
0
0
0
1
(.02973)
1/2
(.01635)
1/2
(.01107)
1/2
/.239
/.239
/.239
=
.
.
7214
5350
.
4402
.
Application of Rigid Body Parameters to Bond Length Corrections
One of the most important applications of the analysis of thermal parameters, and especially of rigid body parameters, is to correct observed bond lengths for thermal motion effects. By the phrase, “observed bond lengths”, it is meant the bond lengths computed from the refined positions of the atoms. It is well known that the observed bond lengths may not represent the true interatomic separations, but to determine the true lengths requires an understanding of the correlation in the motions of bonded atoms.
Busing and Levy (1964b) outlined several models for correcting bond lengths. The most famous is the “riding model” where one of the atoms is assumed to be strongly bonded to a much heavier atom, upon which it “rides”, such as an OH bond. There has been quite a bit of research on how to correct the bond lengths for thermal motion effects
(Cruickshank, 1956; Busing and Levy, 1964b; Schomaker and Trueblood, 1968; Johnson,
1969a,b; Dunitz et al, 1988) and these have been reviewed by Downs et al (1992).
However, in practice it is quite difficult to determine the correlation in the motions of the bonded atoms. Since the various models produce various corrected bond lengths, the end result is that researchers seldom try to compute the true bond length. For instance, see
Winter et al (1977) where they attempt to correct bond lengths obtained for low albite as a function of temperature.
One exception is the rigid body model. If a group of atoms satisfies the rigid body criteria then the assumption of rigid body correlation in the motion of a pair of atoms can be justified. Schomaker and Trueblood (1968) and Johnson (1969b) derive an expression that can be used to correct the bond vector between coordinated pairs of atoms. If [ v ]
C
is the bond vector expressed in the Cartesian system used to solve the TLS model, then the correction to this vector, [ ∆ v ]
C
, is found by
[ ∆ v ]
C
= ½ [trace(L)I
3
– L][ v ]
C
, where I
3
is the 3 × 3 identity matrix. The derivation is somewhat involved, and so will not be presented here.
Example . Compute the observed and corrected SiO bond length for β quartz at 848 K
(Kihara, 1990). In the Cartesian system used to solve the TLS problem, the interatomic vector from Si to O is [-.94234, .89831, .90950]. Therefore, the observed bond length is
(-.94234
2 + .89831
2 + .90950) 1/2 = 1.588 Å, and
[ ∆ v ]
C
= ½ [trace(L)I
3
– L][ v ]
C
= ½ [0.05715
1
0
0
0
1
0
0
0
1
-
−
=
0 .
.
.
02584
01833
02096
0 .
02973
0
0
0
0 .
01635
0 0
0
0
.
01107
]
−
0
0 .
.
.
94234
89831
90950
24
The corrected vector is then [ v ]
C
+ [ ∆ v ]
C
= [-.95526, 0.91663, 0.93045], and the corrected bond length is (-.95526
2 + 0.91663
2 + 0.93045
2 ) 1/2 = 1.618 Å.
It is not a simple task to compute bond lengths corrected for rigid body motion because of the fitting procedures and so on. Consequently, Downs et al (1992) derived a model for the “simple rigid bond” for rigid coordinated polyhedra based upon the assumption that the central cation only undergoes translational motion,
R 2
SRB
= R 2 obs
+
3
8 π 2
(B eq
(Y) – B eq
(X)), where X is the central cation and Y is the anion. This model reproduces the TLS corrected bond lengths very well, and can be applied to data with only isotropic temperature factors, such as those obtained at high pressures.
Example . Compute the simple rigid bond corrected SiO bond length for β quartz at 848
K (Kihara, 1990). B eq
(Si) = 1.81 Å 2 and B eq
(O) = 4.33 Å 2 , and R obs
= 1.5881 Å, so
R 2
SRB
= 1.5881
2 +
3
8 π 2
(4.33 – 1.81) = 2.6178, and the corrected SiO bond length is 1.618 Å.
All the SiO bond lengths in both α and β quartz were corrected for thermal motion effects using the TLS and SRB models and are given in Table 2.
Table 2. The observed and corrected SiO bond lengths for quartz (Kihara,
1990) as a function of temperature. The bond lengths for α quartz have been averaged.
T (K) R obs
(SiO) (Å) R
TLS
(SiO) (Å) R
SRB
(SiO) (Å)
α -quartz
1.609 1.615 1.615
398 1.608 1.615 1.615
498 1.607 1.616 1.616
597 1.605 1.616 1.616
697 1.602 1.616 1.616
773 1.600 1.616 1.616
813 1.597 1.615 1.616
838 1.594 1.616 1.616
β -quartz
848 1.588 1.618 1.618
854 1.588 1.618 1.618
859 1.588 1.618 1.618
869 1.588 1.618 1.618
891 1.588 1.618 1.618
920 1.589 1.618 1.619
972 1.588 1.618 1.618
1012 1.588 1.619 1.619
25
1078 1.587 1.619 1.619
The corrected and observed bond lengths are plotted in Figure 5. The observed bond lengths in quartz as a function of temperature are physically unrealistic in that they decrease with increasing temperature. However, the corrected bond lengths slowly increase in a systematic way, with R corr
(SiO) = 1.6144(4) + 2.3(6) × 10 -6 T for α quartz and
R corr
(SiO) = 1.612(1) + 7(1) × 10 -6 T for β quartz.
1.63
1.62
Corrected
1.61
1.60
Observed
1.59
1.58
200 400 600
Temperature (K)
800 1000
Figure 5 . The variation of the observed and thermally corrected SiO bond lengths in α and β quartz as a function of temperature. The observed data are platted as circles and the corrected data are plotted as squares. The two non-equivalent bond lengths are averaged in α quartz. The observed data decrease systematically in α quartz and are relatively constant in β quartz.
The corrected data increase systematically over the whole temperature range for both α and β quartz.
REFERENCES
Bartelmehs, K.L. (1993) Modeling the properties of silicates. Dissertation submitted to the Faculty of the Virginia Polytechnic Institute and State University, Blacksburg,
Virginia, 181 pp.
Bartelmehs, K.L., Downs, R.T., Gibbs, G.V., Boisen, M.B., Jr., and Birch, J.B. (1995)
Tetrahedral rigid-body motion in silicates. American Mineralogist, 80, 680-690.
Boisen, M.B., Jr. and Gibbs, G.V. (1990) Mathematical Crystallography, Revised
Edition, Volume 15, Reviews in Mineralogy, Mineralogical Society of America,
Washington D.C.
26
Boisen, M.B., Jr. and Gibbs, G.V. (1993) A modeling of the structure and compressibility of quartz with a molecular potential and its transferability to cristobalite and coesite.
Physics and Chemistry of Minerals 20, 123-135.
Born, M. and Huang, K. (1954) Dynamical Theory of Crystal Lattices. Oxford University
Press, Oxford.
Brenner, H. (1967) Coupling between the translational and rotational Brownian motion of rigid particles of arbitrary shape II. General theory. Journal of Colloid and Interface
Science 23, 407-436.
Bürgi, H.B. (1989) Interpretation of atomic displacement parameters: Intramolecular translational oscillation and rigid-body motion. Acta Crystallographica B45, 383-390.
Busing, W.R. and Levy, H.A. (1958) Determination of the principal axes of the anisotropic temperature factor. Acta Crystallographica 11, 450-451.
Busing, W.R. and Levy, H.A. (1964a) The effect of thermal motion on the estimation of bond lengths from diffraction measurements. Acta Crystallographica 17, 142-146.
Busing, W.R. and Levy, H.A. (1964b) The effect of thermal motion on the estimation of bond lengths from diffraction measurements. Acta Crystallographica 17, 142-146.
Busing, W.R., Martin, K.O. and Levy, H.A. (1964) ORFFE, a Fortran function and error program. ORNL-TM-306, Oak Ridge National Laboratory.
Carpenter, G.B. (1969) Principles of crystal structure determination. W.A. Benjamin,
Inc., New York, New York.
Cruickshank, D.W.J. (1956) The analysis of the anisotropic thermal motion of molecules in crystals. Acta Crystallographica 9, 754-756.
Dove, M.T, (1993) Introduction to Lattice Dynamics. Cambridge topics in Mineral
Physics and Chemistry, Cambridge University Press, Cambridge, England.
Downs, R.T., Finger, L.W. and Hazen, R.M. (1994) Rigid body refinement of the structure of quartz as a function of temperature. Geological Society of America
Annual Meeting Abstracts with Programs, 26, A-111.
Downs, R.T., Gibbs, G.V., Bartelmehs, K.L. and Boisen, Jr. M.B. (1992) Variations of bond lengths and volumes of silicate tetrahedra with temperature. American
Mineralogist, 77, 751-757 .
Downs, R.T., Gibbs, G.V. and Boisen, Jr. M.B. (1990) A study of the mean-square displacement amplitudes of Si, Al and O atoms in framework structures: Evidence for rigid bonds, order, twinning and stacking faults. American Mineralogist, 75, 1253-
1267.
Dunitz, J.D., Schomaker, V. and Trueblood, K.N. (1988) Interpretation of atomic displacement parameters from diffraction studies of crystals. The Journal of Physical
Chemistry, 92, 856-867.
Feynman, R.P., Leighton, R.B. and Sands, M. (1977) The Feynman lectures on physics.
Addison-Wesley Publishing Company, Reading, Massachusetts.
Finger, L.W. (1975) Least-squares refinement of the rigid-body motion parameters of
CO
3
in calcite and magnesite and correlations with lattice vibrations. Carnegie
Institution of Washington Year Book, 74, 572-575.
Finger, L.W., and Prince, E. (1975) A system of FORTRAN IV computer programs for crystal structure computations. U.S. National Bureau of Standards Technical Note
854.
Franklin, J.N. (1968) Matrix Theory. Prentice-Hall, Inc. Englewood Cliffs, New Jersey.
27
Ghose, S., Schomaker, V. and McMullan, R.K. (1986) Enstatite, Mg
2
Si
2
O
6
: A neutron diffraction refinement of the crystal structure and a rigid-body analysis of the thermal vibration. Zeitschrift für Kristallographie, 176, 159-175.
Hamilton, W.C. (1959) On the isotropic temperature factor equivalent to a given anisotropic temperature factor. Acta Crystallographica 12, 609-610.
Hirshfeld, F.L. 1976 Can x-ray data distinguish bonding effects from vibrational smearing? Acta Crystallographica A32, 239-244
Hummel, W., Hauser, J., Bürgi, H.-B. (1990) PEANUT: Computer graphics program to represent atomic displacement parameters. Journal of Molecular Graphics, 8, 214-
220.
Hummel, W., Raselli, A., and Bürgi, H.-B. (1990) Analysis of atomic displacement parameters and molecular motion in crystals. Acta Crystallographica, B46, 683-692.
Jacobsen, S.D., Smyth, J.R., Swope, R.J., and Downs, R.T. (1999) Refinement of the crystal structures of celestite (SrSO body character of the SO
4
4
), anglesite (PbSO
4
), and barite (BaSO
groups. The Canadian Mineralogist, in press.
4
): Rigid-
Johnson, C.K. (1969a) An introduction to thermal-motion analysis. In Crystallographic
Computing, editor F.R. Ahmed. Munksgaard, Copenhagen.
Johnson, C.K. (1969b) The effect of thermal motion on interatomic distances and angles.
In Crystallographic Computing, editor F.R. Ahmed. Munksgaard, Copenhagen.
Johnson, C.K. (1970) Generalized treatments for thermal motion. In Thermal Neutron
Diffraction. B.T.M. Willis, editor. Oxford University Press.
Kihara, K. (1990) An X-ray study of the temperature dependence of the quartz structure.
European Journal of Mineralogy 2, 63-77.
Kunz, M. and Armbruster, T. (1990) Difference displacement parameters in alkali feldspars: Effect of (Si,Al) order-disorder. American Mineralogist, 75, 141-149.
Landau, L.D., and Lifshitz, E.M. (1988) Mechanics. Pergamon Press, Elmsford, New
York.
Lipson, H. and Cochran, W. (1953) The determination of crystal structures. G. Bell and
Sons Ltd. London, England.
Markgraf, S.A. and Reeder, R.J. (1985) High-temperature structure refinements of calcite and magnesite. American Mineralogist, 70, 590-600.
Nelmes, R.J. (1969) Representational surfaces for thermal motion. Acta
Crystallographica A25, 523-526.
Peterse, W.J.A.M. and Palm, J.H. (1966) The anisotropic temperature factor of atoms in special positions. Acta Crystallographica 20, 147-150.
Pilati, T., Demartin, F. and Gramaccioli, C.M. (1994) Thermal parameters for α -quartz:
A lattice-dynamical calculation. Acta Crystallographica B50, 544-549.
Schomaker, V. and Trueblood, K.N. (1968) On the rigid-body motion of molecules in crystals. Acta Crystallographica B24, 63-76.
Stuckenschmidt, E., Joswig, W. and Baur, W.H. (1993) Natrolite, Part I: Refinement of high-order data, separation of internal and external vibrational amplitudes from displacement parameters. Physics and Chemistry of Minerals, 19, 562-570.
Trueblood, K.N. (1978) Analysis of molecular motion with allowance for intramolecular torsion. Acta Crystallographica A34, 950-954.
Waser, J. (1955) The anisotropic temperature factor in triclinic coordinates. Acta
Crystallographica 8, 731.
28
Willis, B.T.M. and Pryor, A.W. (1975) Thermal Vibrations in Crystallography.
Cambridge University Press. Cambridge, England.
Winter, J.K., Ghose, S. and Okamura, F.P. (1977) A high-temperature study of the thermal expansion and the anisotropy of the sodium atom in low albite American
Mineralogist, 62, 921-931.
29