4 Phenols C
advertisement

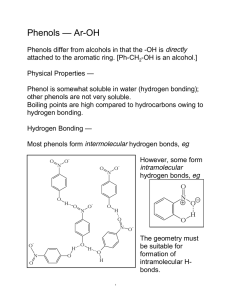
CHAPTER 4 Phenols 4.1 INTRODUCTION Phenols are the compounds containing a hydroxyl group (—OH) directly attached to an aromatic ring. The term phenol is commonly used in the context of hydroxybenzene, which is a liquid at room temperature when contaminated with a little water. It is the carbolic acid of pharmacy. It was the first chemical to be used as an antiseptic as early as 1867 (Lister). It is being used as an important raw material in the manufacture of synthetic polymers (plastics). A number of phenols and phenolic ethers occur in nature. Salicylic acid, for example, occurs in willow tree. Some other important salicylic acid derivatives are methyl salicylate or oil of wintergreen —a common ingradient of liniments, and acetylsalicylic acid (aspirin)—a time honoured analgesic and antipyretic drug. Thymol is a typical flavouring ingredient of thyme and is widely used in the preparation of mouthwash because of its flavour and antiseptic property. Clove oil, used by dentists as an antiseptic, also contains a phenol, eugenol. In addition, certain phenolic compounds are known for their specific physiological actions. For instance, poison ivy (irritants) are 1,2-dihydroxybenzene derivatives having a long side-chain at 3-position. A complex phenolic compound, tetrahydrocannabinol, is one of the active principles of the intoxicant marijuana. OH OH OH COOH Phenol (Carbolic acid) OCOCH3 Salicylic acid (a phenol and an acid) CH3 COOH3 Methyl salicylate (a phenolic ester) OH OCH3 COOH Acetylsalicylic acid (Aspirin) OH CH(CH3)2 Eugenol 146 CH2CH Thymol CH2 PHENOLS 147 CH3 OH OH OH OCH3 OH H O C15 side chain Poison ivy irritants n-C5H11 CHO Vanillin CH3 CH3 (–)-Tetrahydrocannabinol CLASSIFICATION AND NOMENCLATURE We have mentioned above that phenols are hydroxy derivatives of aromatic (benzenoid) compounds. They are represented by the general formula Ar—OH. Depending upon the number of hydroxy groups, they are classified as monohydric (one—OH group), dihydric (two—OH groups), trihydric (three—OH groups) and polyhydric (more than three—OH groups) phenols. Following two systems are in use for naming these compounds: (i) Common system: A number of phenols are assigned special names, while others are named as derivatives of these substances. Compounds having only one additional substituent are named as the derivatives of phenol, the position of this substituent is indicated by letters o-, m-, or p-. Some examples are given below: Monohydric phenols OH OH OH OH CH3 CH3 CH3 Phenol o-Cresol p-Cresol m-Cresol Dihydric phenols OH OH OH OH OH OH Catechol Resorcinol Quinol (Hydroquinone) Trihydric phenols OH OH OH HO OH Phloroglucinol OH Pyrogallol Chapter 4 4.2 H 148 ORGANIC CHEMISTRY [VOL-II] (ii) IUPAC system: In this system the simplest phenol is called benzenol. But all substituted phenols are named as derivatives of phenol. The carbon atoms of the aromatic ring are numbered commencing with the carbon atom bearing the root functionality (the —OH groups), the ring carbons are numbered successively so that the sum of numbers used to designate the position of substituents is minimum. Following examples are illustrative (common names in parentheses). OH OH OH OH CH3 CH3 Benzenol (Phenol ) 2-Methylphenol (o-Cresol) NH2 3-Methylphenol (m-Cresol) 3-Aminophenol (m-Aminophenol) Dihydric, trihydric and polyhydric phenols are named as benzenediols, benzenetriols and benzenepolyols, respectively. These names are also written as hydroxy derivatives of benzene. However, they are better known by their trivial names parentheses. For instance, OH OH 1 1 2 OH OH 1 2 3 1, 2-Benzenediol or 1, 2-Dihydroxybenzene (Catechol ) 2 6 OH HO 1, 3-Benzenediol or 1, 3-Dihydroxybenzene (Resorcinol) 5 4 3 OH 1, 3, 5-Benzenetriol or 1, 3, 5-Trihydroxybenzene (Phloroglucinol) OH OH 1 OH 2 2 HO 3 1 3 4 OH 1,2,3-Benzenetriol or 1,2,3-Trihydroxybenzene (Pyrogallol) OH 1,2,4-Benzenetriol or 1,2,4-Trihydroxybenzene (Hydroxyquinol) Phenols with other substituents OH 1 6 5 OH 2 Cl 3 4 1 6 5 4 2 NO2 3 Cl Cl 2, 4-Dichlorophenol 4-Chloro-2-nitrophenol However, when a functional group such as carboxylic group, ester or carbonyl group is present in addition to phenolic group, the phenols are named as hydroxy derivatives of these compounds. The common names of these compounds are retained as root names. The ring is numbered commencing with the designated functional group and going round successively as above. Thus, PHENOLS 149 CHO 1 6 5 Cl CHO 1 OH 2 6 5 3 OH OH 5-Chloro-2, 4-dihydroxybenzaldehyde 4-Hydroxy-2-nitrobenzaldehyde COOH 1 6 5 COOC2H5 1 OH 2 6 5 3 4 4 OH 2 Cl 3 OH 2, 4-Dihydroxybenzoic acid (β-Resorcylic acid) 4.3 NO2 3 4 4 2 Ethyl 2-chloro-4-hydroxybenzoate STRUCTURE AND BONDING sp 3 sp 3 sp 3 H 1s O sp 3-1s sp 3 sp 2 sp2 –sp3 C O, σ - bond H O 136 pm 109° Fig. 4.1: Structure of phenol Due to higher electronegativity of oxygen atom, phenol molecule is dipolar in nature with the oxygen carrying partial negative charge. Due to this dipolar nature phenols form hydrogen bonds. It is noteworthy that the dipole moment of phenol (1.54D) is smaller than that of methanol (1.71D), because the C—O bond in phenol is less polar due to electron-withdrawing effect of the benzene ring while in methanol, C—O bond is more polar due to electron-donating effect of methyl group. The phenol molecule cannot be depicted by any single valence bond structure. In fact, it is considered as a hybrid of the following contributing forms: Chapter 4 2 In phenols, the C—O bond is formed by the overlap of sp -orbital of carbon of benzene ring with 3 a sp -orbital of oxygen atom while the O—H bond is formed by the overlap of second 3 sp -orbital of oxygen with 1s orbital of hydrogen (Fig 4.1). The remaining two non-bonding sp3-orbitals of oxygen atom contain lone-pairs of electrons. 150 ORGANIC CHEMISTRY [VOL-II] H + O H + O H H + O O OH – – – I III II IV V Resonating structures of phenol A perusal of the contributing forms II, III and IV clearly shows that the oxygen atom acquires a positive charge due to resonance. This polarity facilitates release of proton and formation of phenoxide ion which is also stabilized by resonance. – O O O O O – – – – Hydrogen bonding in phenols (i) Intermolecular hydrogen bonding: Like other compounds having —OH groups phenols either solids or liquids, exhibit intermolecular hydrogen bonding. Due to these hydrogen bonds they exist as polymeric aggregates held together. These aggregates break up on dilution with a non-polar solvent first into trimers or dimers and finally into the monomers on large dilution. H H O H O H O H O O It has been observed that the boiling point of phenol is higher as compared to toluene (having comparable mol. weight). This is due to the formation of intermolecular hydrogen bonding which results in the formation of polymeric aggregate where molecular mass increases many fold, thereby raising its boiling point. The reason for the high boiling point may be due to the fact that additional energy is required to break the hydrogen bonds. Toluene, on the other hand, does not form hydrogen bonds. Phenol is somewhat soluble in water because it forms cross-intermolecular hydrogen bonding with water molecules, as shown below: H O H O H H O H O H H O H O H (ii) Intramolecular hydrogen bonding: A phenol, in which a carbonyl or a nitro group is attached at the ortho position, usually forms intramolecular hydrogen bonding as shown below: PHENOLS 151 CH3 O C N O O + O H O – H Intramolecular hydrogen bonding in o-Hydroxyacetophenone and o-Nitrophenol This type of hydrogen bonding also alters the physical and chemical properties of these molecules. 4.4 METHODS OF FORMATION (i) From coal-tar: The middle oil fraction (443–513K) of coal-tar consists of phenol and cresols in addition to other compounds. This fraction is collected and treated with alkali. The alkaline layer is separated and carbon dioxide gas is bubbled through it. The phenolic mixture that separates out is subjected to fractional distillation in order to isolate individual phenols. (ii) From halobenzenes (Dow process): Chlorine in chlorobenzene is inert to nucleophilic displacement under usual conditions. However, when chlorobenzene is heated with sodium hydroxide at 613K under pressure it forms phenol. This method is used for commercial production of phenol and was first developed by Dow chemicals, USA in 1928. OH Chapter 4 Cl – OH, 613K Pressure (320atm) Mechanism: This transformation takes place via benzyne mechanism as shown below: Cl 8%NaOH 613K, 320atm OH H – H – + OH – OH OH – –OH NaOH ONa –H2O However, if halobenzene has a strong electron-withdrawing substituent at ortho- or para-position, the hydrolysis of these compounds becomes easier. Thus, 2,4-dinitrophenol and 2,4,6-trinitrophenol (picric acid) are obtained from the corresponding aryl halides using milder conditions. Cl NO2 15% NaOH 433K O2N Cl NO2 HO O2N NO2 Aq. Na 2CO 3 403K HO NO2 152 ORGANIC CHEMISTRY [VOL-II] O2 N O2 N NO2 Cl H2O NO2 HO 333K O2N O2 N 2,4,6-Trinitrochlorobenzene (Picryl chloride) 2,4,6-Trinitrophenol (Picric acid) (iii) From isopropylbenzene (cumene): This procedure is essentially used for the preparation of the parent compound, phenol. Cumene (A) on catalytic aerial oxidation gives phenol via cumene hydroperoxide (B) CH3 CH3 C 6 H5 C O2 Catalyst H C 6 H5 C O CH3 CH3 (A) (B) O – CH 3COCH 3 H C6H5OH Phenol This reaction, which is an example of autooxidation, is carried out at 423K in presence of hydrogen bromide as a catalyst. It takes place by a free radical chain mechanism and is initiated by the abstraction of hydrogen from hydrocarbon by a bromine radical, which is produced by aerial oxidation of H—Br. The free hydrocarbon radical thus produced picks up a molecule of oxygen forming peroxy radical which in turn abstracts hydrogen atom from H—Br in the propagation step to give cumyl hydroperoxide. Initiation H Br + O2 Br + HOO Hydroperoxide radical CH3 C6H5 CH3 C H + Br C6H5 CH3 C + HBr CH3 Propagation CH3 CH3 C + O2 C6H5 C6H5 C CH3 C CH3 O CH3 CH3 C6H5 O CH3 O O + HBr C6H5 C CH3 O OH + Br PHENOLS 153 The cumyl hydroperoxide so obtained is treated with acid to form phenol. The reaction is called cumyl hydroperoxide rearrangement. CH3 + C6H5 C O H OH C6H5OH + CH3 CO CH3 CH3 The mechanistic path of this rearrangement involves initial protonation of the —OH group of hydroperoxide. The resulting oxonium ion loses water to form a species with electron-deficient oxygen. A carbocation, generated by phenyl migration, is stabilized by resonance. Nucleophilic attack of water on this carbocation gives a hemilketal which undergoes acid-catalysed split to form the products. CH3 CH3 C O O H + + + H C6H5 CH3 O OH2 CH3 CH3 + C6H5 C O C CH3 1, 2-Phenyl shift C6H5 CH3 C O + CH3 + C6H5 O C CH3 + OH2 H2 O CH3 C6H5 + OH C6H5 O C CH3 CH3 O C CH3 CH3 CH3 + H (A hemiketal) C6H5 O –H H + CO C6H5 O H O C CH3 H CH3 + + –H CH3 (iv) From diazonium salts: Addition of diazonium salt solution to a large excess of warm 50% sulphuric acid at 323K results in the formation of phenol. + Ar N – NX + H2O + H 323 K Ar OH + N2 + HX Chapter 4 C6H5 154 ORGANIC CHEMISTRY [VOL-II] (v) From sulphonic acids: Fusion of the alkali metal salt of an aromatic sulphonic acid with sodium or potassium hydroxide (solid) affords the corresponding phenol. – + ArSO3Na + NaOH fuse (573 K) –Na2SO 3 – ArO + +H ArOH Phenoxide This reaction is of general utility. However, the hydrolysis of diazonium salt solution is a preferable procedure as the fusion carried out at higher temperatures leads to undesired side products. 4.5 PHYSICAL PROPERTIES Various physical parameters such as boiling points, melting points, water solubilities and Ka values of some phenols listed in Table 4.1, are discussed below: (i) Physical state: A look at the Table reveals that most of the simpler monohydric phenols are either liquids or low melting solids. Pure phenols are colourless but they usually turn reddish brown due to atmospheric oxidation. (ii) Melting and boiling points: Nitrophenols, aminophenols and phenols having more than one hydroxyl group have relatively higher melting and boiling points. This is probably due to the increased polar character resulting in higher degree of association involving intermolecular hydrogen bonding. In general phenols are more polar than cycloalkanols having similar carbon skeletons. This difference in polarities is reflected in higher melting and boiling points of phenol (m.p. 316K; b.p. 454K) as compared to those of cyclohexanol (m.p. 298K; b.p. 434K). Table 4.1: Physical Properties of Some Phenols Name phenol o-cresol m-cresol p-cresol o-fluorophenol m-fluorophenol p-fluorophenol o-aminophenol m-aminophenol p-aminophenol o-nitrophenol m-nitrophenol p-nitrophenol 2, 4-dinitrophenol 2, 4, 6-trinitrophenol catechol resorcinol m.p. (K) 316 303.5 284 309 289.1 287 321 447 396 459 318 369 387 386 395 377 383 b.p. (K) 454 463 473 474 425 451 459 — — — 490 — — — — — — Ka(H2O) at 298 Water solubility × 10–10 g/100g of H2O 1.3 0.63 0.9 0.62 1.5 5.2 1.1 2.0 69 — 600 50 690 1000000 very large 1.0 3.0 9.3 2.5 2.6 2.4 — — — 1.7 2.6 1.0 0.26 1.45 1.72 1.6 1.4 1.45 123 PHENOLS 155 hydroquinone pyrogallol 1, 2, 4-trihydroxybenzene phloroglucinol 446 406 413 490 — — — — 2.0 1.0 3.0 — 8 62 1.1 — (iii) Solubility: The solubility of phenol (m.w. – 94, 9.3 g/100 g of H2O) is more than that of the cyclohexanol (m. w. – 100, 3.6 g/100 g of H2O). This difference can be attributed to the fact that the δ– δ+ phenolic —OH group is more polarized (as —O —H ) than alcoholic —OH group because of the resonance involving benzene ring in the case of the former. Further, among the isomeric fluoro- and nitrophenols, the ortho isomers have lower melting points, boiling points and water solubilities and are weaker acids than the corresponding meta and para-isomers. This is due to intramolecular hydrogen bonding in the case of ortho isomers and intermolecular hydrogen bonding in the case of meta- and para-isomers. O O H – H o-Nitrophenol (Intramolecular hydrogen bonding) 4.6 O N + O – H O N O – + p-Nitrophenol (Intermolecular hydrogen bonding) SPECTRAL CHARACTERISTICS (i) Infrared spectra: Like alcohols the infrared spectra of phenols are characterized by the typical band in the 3600–3200 cm–1 region due to O—H stretching vibrations. This band, however, shifts to 3610 cm–1 on dilution, due to the ‘free’ O—H group. Further, the O—H stretching vibrations of those phenols which are capable of forming intramolecular hydrogen bonding appear in the 3200–2500 cm–1 region. The phenols are distinguished from alcohols because of different frequencies of C—O stretching vibrations which show up at 1230 cm–1 in the former (Fig. 4.2). The other characteristics of the infrared spectra are the typical absorption bands as expected from benzene derivatives. μm 2.5 15 OH 3040 1465 1580 3340 4000 1492 cm–1 Fig. 4.2: The IR spectrum of phenol 1230 800 Chapter 4 O O O N 156 ORGANIC CHEMISTRY [VOL-II] (ii) Ultraviolet spectra: The ultraviolet spectra of phenols are characterized by the typical E and B bands of aromatics which appear at relatively longer wavelength compared to benzene (bathochromic shift). This is probably due to the extended conjugation involving —OH group. The λmax of phenol, for instance, appears at 215 nm, 270 nm and 275 nm. Additional bathochromic shift appears in alkaline solution because of resonance involving phenoxide ion (dispersal of charge). 4.7 CHEMICAL REACTIONS (i) Acidic character: Both alcohols and phenols contain an —OH group and due to difference in electrongativities of oxygen and hydrogen, both types of compounds are expected to be acidic in nature. Though both exhibit acidic properties (react with electropositive metals), phenols are considerably more acidic than alcohols. This difference in acid strength is reflected in the formation of phenoxide salts when phenols are treated with aqueous alkali whereas alcohols do not react under these conditions. R R – O R O H + O – O + H2O H – H + O R H – O + H2O Explanation for greater acid strength of phenols than alcohols. Alcohols neither react with NaOH nor turn blue litmus red. This could be explained by considering the fact that due to resonance involving benzene rings, phenols and phenoxides are more stable than alcohols and alkoxides, respectively. Further, as shown in Fig. 4.3, the phenoxide ions are much more stable than alkoxides ions due to dispersal of negative charge in the former, whereas the difference in stability of phenols (involving separation of charge) and alcohols is not much pronounced. O H + O H + O H + O H OH – – – Resonance stabilization of phenol (separation of charge, less stable) – O O O – O O – – – Resonance stabilization of phenoxide ion (dispersal of charge, more stable) PHENOLS 157 Thus acid strength of phenol becomes evident from the fact that phenoxide ion is resonance stabilized to a larger extent due to dispersal of charge, compared to phenol where resonance involves separation of charge. – Extent of resonance stabilization of phenoxide compared to alkoxide RO – ArO Extent of resonance stabilization of phenol compared to alcohol ROH ArOH Comparison of acid character of phenols and carboxylic acids Though stronger than alcohols, phenols are weaker acids than carboxylic acids (Ka = 10–5) or even carbonic acid (Ka= 10–7). This is the reason why phenols fail to react with sodium carbonate or bicarbonate. In fact the phenols are precipitated from aqueous solution of phenoxides by bubbling carbon dioxide gas. Further, the difference in acid strength of phenols and carboxylic acids provides a convenient method for their separation with aqueous sodium bicarbonate in which the latter are soluble leaving the former behind. Effect of substituents on the acid strength of phenols The acid strength of phenols is effected considerably by the presence of substituents on the ring. An electron-withdrawing substituent helps in greater dispersal of negative charge on the phenoxide ion either by inductive effect (–I) or by resonance effect (–R) or both. This would result in the increase in acid strength of phenol. Conversely, the electron-donating groups due to +I or +R or both effects, decrease the acid strength. (i) Effect of electron-withdrawing substituents: Phenols having electron-withdrawing groups (–I, –R) such as —CN, —NO2, —COOH, —CHO, —COR etc., at ortho and para-positions with respect to the phenolic group are stronger acids than phenol. Here it may be mentioned that only the inductive effect is not responsible for the increase in acid strength. In fact it is the resonance effect (–R in this case) which plays a major role in stabilising the corresponding phenoxide ion. Since the conjugation is extended upto oxygen of the nitro group, this anion is more stable than phenoxide ion where no such extended conjugation is possible. This is evident from the fact that p-nitrophenol (Ka = 700 × 10–10) is 700 times as strong as phenol (Ka = 1 × 10–10). –O O O O O– O – – – NO2 NO2 N – – + O O O NO2 N + O – Resonance stabilization of p-nitrophenoxide ion NO2 Chapter 4 Fig. 4.3: Comparative acidic strength of phenols and alcohols 158 ORGANIC CHEMISTRY [VOL-II] As the number of contributing forms is more and the negative charge is spread over two electronegative oxygen atoms, the resulting anion is more stabilized than phenoxide ion. (ii) Effect of electron-donating substituents: Phenols having electron donating groups (+I, +R) such as —CH3, —OCH3, —NH2., etc., at ortho or para position with respect to —OH group are weaker acids.This may be attributed to the intensification of negative charge on the corresponding phenoxide ion due to resonance (+R), resulting in its destabilization. – O CH3 Charge on oxygen intensified (less stable than phenoxide) However, the groups such as —NH2, —OH, —OR etc., when present in m-position increase the acid strength of phenols due to –I effect. The order of acid strength for some typical phenols is as under: OH OH > OH OH > NO2 > > CH3 Cl p-Nitrophenol OH p-Chlorophenol Phenol OCH3 p-Cresol p-Methoxyphenol Thus, in nutshell, electron-withdrawing substituents increase the acid strength of phenols while electron-donating substituents decrease their acid strength. (iii) Effect of position of the substituent: It may be worthwhile to understand here that the acidweakening effect of the electron-donating groups and acid-strengthening effect of the electronwithdrawing groups is more pronounced at o- and p-positions with respect to the OH group than at m-position. This may be explained on the basis of the fact that both inductive and resonance effects influence the stability of phenoxide ion left after the removal of a proton. In some cases, both inductive and resonance effects reinforce while in other cases they may oppose each other depending upon the nature and position of the substituent on the benzene ring as discussed below: (a) Resonance effects: Nitro group has a powerful –I as well as –R-effect, therefore, irrespective of the position of nitro group, all nitrophenols are stronger acids than phenol. Although –I-effect decreases with distance, –R-effect is more pronounced at o-and p-positions than at m-position. To explain this, let us compare the stabilities of o-, m- and p-nitrophenoxide ions. – O O NO2 O O + NO2 – – N O – – O O + N O – O O – O O + + N N – O– Resonance stabilization of o-nitrophenoxide ion I O – PHENOLS – 159 O O O O O O – – – – NO2 NO2 – N N + + O – O O NO2 NO2 O – II Resonance stabilization of p-nitrophenoxide ion – O O O O – – – NO2 O NO2 NO2 – NO2 NO2 It is clear from the above structures that both o- and p-nitrophenoxide ions are stabilized by five resonating structures. In one of the structures in each case (I or II), conjugation is extended upto oxygen atom of the nitro group. But no such conjugation is possible for m-nitrophenoxide ion due to which m-nitrophenoxide is stabilized by only four resonating structures. In other words, o-nitrophenoxide ion and p-nitrophenoxide ions are more stable than m-nitrophenoxide ion. Therefore, o-nitrophenol and p-nitrophenol are stronger acids than m-nitrophenol. Out of o-nitrophenol and p-nitrophenol, o-nitrophenol is little less acidic than p-nitrophenol. This may be due to the fact that acidic hydrogen of OH group is involved in intramolecular H-bonding or chelation which makes loss of the proton a little more difficult. O + O N – H O Thus, the acid strength of nitrophenols relative to phenol decreases in the order: p-Nitrophenol > o-Nitrophenol > m-Nitrophenol > Phenol Further, greater the number of electron withdrawing groups at o- and p-positions, more stable is the phenoxide ion and hence more acidic is the phenol. Thus, acid strength of nitrophenols with respect to phenol decreases in the order: 2,4,6-Trinitrophenol > 2,4-Dinitrophenol > 4-Nitrophenol or 2-Nitrophenol > Phenol (b) Inductive and hyperconjugation effects (i) Comparison of the acidic strength of halophenols. Halogens have +R and –I-effects, but the –I-effect predominates over the +R-effect. Therefore, all halophenols (except p-fluorophenol) are more acidic than phenol itself. Further, since –I-effect decreases with distances, the acidic strength of halophenols decreases in the order: o-Halophenol > m-Halophenol > p-Halophenol Chapter 4 Resonance stabilization of m-nitrophenoxide ion 160 ORGANIC CHEMISTRY [VOL-II] In case of p-fluorophenol +R-effect and –I-effect of F almost balance each other due to almost identical sizes of 2 p-orbitals of C and F and hence it is almost as acidic as phenol itself. Out of o-halophenols, o-fluorophenol is the weakest acid due to strong intramolecular H-bonding while the acid strength of other halophenols decreases as the —I-effect of the halogen decreases. F H O Therefore, the acid strength of all the o-halophenols decreases in the order: o-Chorophenol > o-Bromophenol > o-Iodophenol > o-Fluorophenol (ii) Comparison of the acidic strength of cresols. The alkyl groups are electron donating due to hyperconjugation effect. Therefore, all cresols (methylphenols) are less acidic than phenol itself. Futher, hyperconjugation effect cannot operate at m-position due to which m-cresol is more acidic than o- and p-cresols. Due to field effects which make the loss of a proton little more difficult, p-cresol is more acidic than o-cresol. Thus, the acid strength of cresols relative to phenol decreases in the order: phenol > m-cresol > p-cresol > o-cresol. (iii) Comparison of acid strength of dihydric phenols i.e, catechol, resorcinol and hydroquinone. In case of catechol due to intramolecular H-bonding the loss of a proton is little difficult compared to that in hydroquinone. Hence hydroquinone is more acidic than catechol. But in case of resorcinol, the two OH groups are situated at m-position due to which one of them can not enter into the resonance with the other OH group. Instead –I-effect of one OH groups on the other makes resorcinol more acidic than catechol and hydroquinone. Thus, the order is: Resorcinol > Hydroquinone > Catechol (iv) Comparison of acid strength of phenol with m-methoxyphenol and m-aminophenol. A group present at m-position cannot enter into resonance with the hydroxy group of phenols, but can exert inductive effect from this position. Due to –I-effect of both methoxy and amino groups, m-methoxyphenols and m-aminophenols are more acidic than phenol. Futher, due to more –I-effect of methoxy group than of amino group, m-methoxyphenol is a stronger acid than m-aminophenol. Therefore, their acid strength decreases in the order: m-methoxyphenol > m-aminophenol > phenol. (iv) Ester formation–Acylation: Phenols are converted into the corresponding esters by the action of acid chlorides or acid anhydrides in presence of either acidic or basic catalysts. Ar OH + CH3 CO X aq. NaOH or pyridine or conc. H2SO4 (X= Cl, OCOCH3) Ar O Ester CO CH3 + HX Different mechanisms operate under different conditions. (a) Base catalysed esterification (BAC 2 mechanism): Under basic conditions the reaction is initiated by the nucleophilic addition of phenoxide anion, (produced by the action of alkali on phenol) on the carbonyl carbon of the acid chloride or anhydride, forming a tetrahedral intermediate. Loss of V X from the intermediate gives rise to the products. PHENOLS 161 – Ar O – H + OH Ar R R – Ar O O + H2O – + C O O Ar C X – O R CO O Ar + X X The reaction of phenol with benzoyl chloride in presence of NaOH is called Schotten-Baumann reaction. OH + Cl CO Phenol NaOH –HCl Benzoyl chloride C6H5 O CO C6H5 Phenyl benzoate H3 C C O H3 C H3 C + H + C X Chapter 4 (b) Acid catalysed esterification (AAC 2 mechanism): The reaction is initiated by protonation of the carbonyl oxygen resulting in the development of full positive charge on carbonyl carbon so that a relatively weak nucleophile such as phenol can attack. Proton exchange followed by elimination of HX gives the product. Acetylation with acidic anhydride in presence of concentrated sulphuric acid takes place in the following steps: OH + C OH Ar OH X X H3 C H3 C + Ar O C H X OH + –H + +H H3C + Ar O C O H Ar O C O + H + HX + XH Fries rearrangement: The phenolic esters, upon heating with anhydrous aluminium chloride, are converted into the isomeric o- and p-hydroxy ketones or more often, into a mixture of both. OCOR OH OH COR AlCl3, Δ + COR The reaction is called Fries migration or Fries rearrangement. It has been shown that the p-isomer predominates at lower temperatures whereas o-isomer is obtained in larger amounts at higher temperatures for obvious reasons (steric effects) and the p-product can often be transformed into o-isomer on further heating with aluminium chloride. 162 ORGANIC CHEMISTRY [VOL-II] OH OCOCH3 COCH3 AlCl3, 298K AlCl3, 438K o-Hydroxyacetophenone HO COCH3 p-Hydroxyacetophenone Phenyl acetate Although p-isomer is obtained more easily, the o-isomer is more stable, due to intramolecular hydrogen bonding. O H O C CH3 Mechanism: The reaction can take place by two alternate mechanistic pathways, i.e., by a one-step or a two-step mechanism. That this reaction takes place by a two-step mechanism in most of the cases, has been proved by the isolation of cross products when a mixture of two identical but differently substituted substrates is treated under the conditions of the reaction. Thus, for instance, esters I and II give the ketones III and IV, respectively. When a mixture of I and II is heated with aluminium chloride, apart from III and IV cross products V and VI are also obtained. OCOC6H5 OH OCOCH3 COC6H5 (i) AlCl3, Δ Cl OH (i) AlCl3, Δ (ii) H+/H2O COCH3 Cl (ii) H+/H2O CH3 CH3 CH3 CH3 I III II IV OH I + II (i) AlCl3, Δ (ii) H+/H2O OH COCH3 III + IV + Cl COC6H5 + CH3 V CH3 VI In the first step an acylium ion (RCO+) is formed which attacks the benzene ring at o- and ppositions in the second step. This two-step mechanism of Fries migration may be outlined as follows: PHENOLS 163 – OCOR – OAlCl3 AlCl3 OAlCl3 + + R CH3 C COR + o-attack O H CH3 OH OAlCl2 COR COR H2 O –HCl CH3 CH3 CH3 One-step mechanism of this reaction, however may be depicted as under: – COR + OCOR – O OAlCl3 COR + AlCl3 OAlCl2 COR H CH3 CH3 OH –HCl COR H2 O CH3 CH3 CH3 (v) Ether formation; Williamson’s synthesis: Phenols can be converted into the corresponding alkyl ethers by treating sodium or potassium phenoxides with alkyl halides. – O + R X ONa + CH3 I Ar – + C6H5 – SN2 O Ar Δ C 6 H5 R + X O CH3 + NaI Alkyl phenyl ethers can also be prepared by treating phenol with dialkylsulphate in weakly alkaline solution. – Ar – O + R O SO2 OR – + C 6 H5 ONa + CH3 O SO2 OCH3 Ar Δ O R + OSO2OR + – C 6 H5 O CH3 + NaOSO2OCH3 Aryl allyl ethers are obtained by action of sodium (or potassium) phenoxide on allyl bromide. – Ar O + Br – CH2 CH CH2 Ar O CH2 CH CH2 + Br Claisen rearrangement: The allyl aryl ethers undergo an interesting transformation, known as Claisen rearrangement when heated at 473 K. This reaction involves migration of an allyl group from ether oxygen to ring carbon at ortho position and when both the ortho positions are blocked, the allyl group migrates to para position. However, in no case does it go to meta position. ortho rearrangement O CH2 CH CH2 473 K OH CH2 CH CH2 Chapter 4 Cl3Al 164 ORGANIC CHEMISTRY [VOL-II] para rearrangement O CH2 H3C CH CH2 CH3 OH 473 K H3C CH3 CH2 CH CH2 Mechanism: The reaction does not require any catalyst and shows first order kinetics with respect to the allyl aryl ether. The Claisen rearrangement is an example of pericyclic reactions and is known as sigmatropic rearrangement. The reaction has been shown to proceed in a concerted manner as evidenced by heating a mixture of ethers VII and VIII having two different allyl groups, whereby cross-products are not obtained. O CH2 CH CHC6H5 OH CH CH CH2 C6H5 VII 473 K + O CH2 CH + CH2 OH CH2 VIII CH CH2 No cross-products are formed. An interesting feature of the rearrangement is that when migration takes place to the ortho position, the γ-carbon of the allyl group (with respect to oxygen) attaches itself to the ring carbon. In other words, ortho migration involves an inversion in the position of substituents with respect to that of the starting compound. However, no such inversion takes place in the case of para migration. ortho rearrangement O CH2 CH CH R OH CH2 473 K CH CH2 R para rearrangement O H3C CH2 CH3 CH CH 473 K R OH H3C CH3 CH2 CH CH R PHENOLS 165 Mechanism of ortho-rearrangement: The above observations point towards the fact that the ortho- isomerization is a concerted process and the reaction proceeds through a six-membered cyclic transition state. The rupture of the allyl-oxygen bond is synchronous with the formation of allylcarbon bond at the ortho position. A cyclohexadienone intermediate IX is thus formed which undergoes prototropic change to give the o-allylphenol. By doing so the ring regains the aromatic character. CH2 CH O O CH CH3 CH2 CH CH slow CH3 Transition state OH O CH CH2 H Prototropic change CH CH3 CH CH2 CH3 IX Mechanism of para-rearrangement: As mentioned above when both the ortho positions are blocked the allyl group migrates to para position without any inversion in the position of substituents on allyl group with respect to the starting compound. This indicates that the overall para migration takes place in two stages. In the first stage the usual ortho migration leading to the formation of cyclohexadienone intermediate takes place. Since there is no hydrogen at this position, tautomerization is not possible hence the ring cannot undergo aromatization. The intermediate, therefore, undergoes another isomerization involving migration of the allyl group to para position again through a sixmembered cyclic transition state with another inversion. Thus, in a para rearrangement one inversion is followed by another and overall there is no inversion. The driving force for the para migration is regaining of aromatic character after allylic migration. CH2 CH O O CH3 H3C CH CH3 CH3 CH3 H3 C CH CH H2 C OH H3 C O Prototropic change CH3 H3 C CH3 H CH2 CH CH CH3 CH2 CH CH CH3 Chapter 4 CH 166 ORGANIC CHEMISTRY [VOL-II] Following observations further confirm the above reaction sequences: (i) The intermediate cyclohexadienone has been trapped by Diels-Alder reaction and has also been synthesized independently. It has been found to give the allyl phenol on heating. (ii) When ortho- 14C-labelled allyl 2,6-diallyl ortho ether was heated the 14C label was found to be equally distributed between ortho and para positions in the rearranged product. This experiment unequivocally established the intermediacy of triallylcyclohexadienone (XII) in which either the labelled or unlabelled allyl group migrates to para position with equal ease. O CH2 CH2 O *CH2 H2 C CH2 CH CH CH CH2 CH2 H2 C CH2 CH CH2 CH CH * 2 CH CH2 XII OH *CH2 H2 C CH2 OH H2 C CH CH CH2 CH + CH2 CH2 CH2 CH CH CH2 CH2 CH CH2 *CH 2 [* = 14C] In addition to aryl alkyl ethers, diaryl ethers of the type Ar—O—Ar’ are also known and they can be obtained by the treatment of a phenoxide with aryl halide at high temperature or in presence of a copper catalyst (Ullmann reaction). The exact role of copper catalyst is not very well understood. It has, however, been proposed that Cu coordinates with halogen and withdraws electrons from C—X bond thus making the displacement of halogen easy. C 6 H5 Br + Cu C 6 H5 – + Br Cu – C6H5O – C 6 H5 O C6H5 + Br + Cu However, if a strong electron-withdrawing group such as a nitro group is present in the orthoor para-position with respect to the halogen atom, it can be displaced by phenoxide anion by a bimolecular nucleophilic displacement without the use of copper catalyst. – O O + – + Cl N O O – – Cl + + O O N O + O – N Cl O – PHENOLS 167 (vi) Electrophilic substitution:The phenolic group or phenoxide anion, because of their ability to accommodate positive charge, are ortho-, para-directing with activation. The activation effect of the OH group is so pronounced that usually polysubstitution occurs. OH OH OH E + + E + + –H E + OH OH + + + E OH OH E E H H OH E E H H + + (More stable) p-attack OH OH + OH OH OH + + + E + H + E H E H E H E (More stable) m-attack OH OH + + E OH + OH + E E H H E + H A look at the carbocations formed by the attack of the electrophile on the ring shows that the carbocations formed during o- and p-attack are resonance hybrids of four contributing forms out of which one form is highly stable because in this each atom (except hydrogen) has a complete octet of electrons. But in case of m-attack the resulting carbocation has no such contributing forms. This Chapter 4 Explanation for ortho, para directing and activating influence of OH group One of the lone pairs of electrons interacts with the π-electrons of the benzene ring due to which the electron density gets increased at all carbon atoms of the ring. This increase in electron density activates the ring for electrophilic substitution. Further, the increase in electron density is higher at ortho- and para-positions relative to meta-position. Therefore, electrophilic substitution is preferred at o- and p-positions in comparison to m-position. This o, p-directing influence of OH group can be explained in terms of the relative stabilities of the carbocations formed during o, p and m-attacks as given below: o-attack 168 ORGANIC CHEMISTRY [VOL-II] indicates that the carbocations formed during o- and p-attack are more stable than those formed by m-attack. Therefore, electrophilic substitutions in phenols occur at o- and p-positions preferentially. Some important electrophilic substitution reactions of phenols are halogenation, nitration, sulphonation and Friedel-Craft reaction which are discussed below: (a) Halogenation: In polar solvents phenols react with halogens (chlorine water or bromine water) at a very fast rate substituting all the available ortho- and para-positions. Thus phenol on treatment with bromine gives 2,4,6-tribromophenol in quantitative yields. This reaction forms the basis of quantitative estimation of phenol. OH OH Br + 3Br2 Br H2O + 3HBr Br 2, 4, 6-Tribromophenol However, halogenation can be stopped at monohalogenation stage if reaction is carried out in presence of non-polar or less polar solvents at low temperature. OH OH Br 2 in CS2 Br OH + 273K Br p-Bromophenol (80%) o-Bromophenol (20%) Reason: It can be explained by considering the fact that in aqueous solution phenol is in equilibrium with phenoxide ion which is the actual substrate and being more reactive than phenol, reacts at a much faster rate. After the substitution the equilibrium is disturbed and in order to restore this equilibrium more phenol is converted into phenoxide which reacts with further amount of bromine. Second and third bromination would take place even at a much faster rate since the presence of electronegative bromine in monobromophenol leads to greater degree of ionization of phenoxide ion. However, in the non-polar solvents such as CCl4 or CS2 this reaction takes place slowly giving rise to mainly the ortho- and para-bromophenol presumably because in such solvents the extent of dissociation of phenol is considerably lower. For similar reasons further bromination is also suppressed. (b) Nitration: Due to their high reactivity, phenols undergo nitration at all available ortho and para positions when treated with conc. nitric acid and sulphuric acid mixture. For instance, phenol gives 2,4,6-trinitrophenol (picric acid) on treatment with conc. nitric acid–sulphuric acid mixture. OH OH HNO 3/H 2SO 4 O2N NO2 Phenol NO2 Picric acid The reaction is very fast but the yields of picric acid are quite low as most of the phenol is oxidized by nitric acid. Picric acid is, therefore, prepared in the laboratory by the following alternative sequence of reactions: PHENOLS 169 (i) From chlorobenzene Cl Cl OH NO2 HNO3/H2SO4 OH NO2 Aq. Na2CO3 HNO3 O2N NO2 NO2 NO2 NO2 (ii) From phenol OH OH OH SO3H Conc. H2SO4 O2N 373K NO2 Conc. HNO3 + Heat SO3H NO2 Picric acid is a very strong acid, being stronger than even carboxylic acids. The presence of three electron-withdrawing nitro groups at the ortho and para positions which are able to accommodate the negative charge of phenoxide ion explains this property. It has been used as a disinfectant and as a reagent in the laboratory for purification of polynuclear aromatic hydrocarbons (PAH) with which it forms well-defined cystalline adducts called picrates. For preparing o- and p-nitrophenols, phenol is treated with dilute nitric acid. The reaction most probably, involves initial nitrosation of phenol with dinitrogen trioxide produced from nitric acid in situ. The nitrosophenol is then oxidized to nitrophenol. OH OH N2O3 OH NO HNO3 NO2 [O] o- and p-Nitrophenol o- and p-Nitrosophenol m-Nitrophenol cannot be prepared by direct nitration of phenol. It may be prepared in good yields according to following reaction sequence commencing with m-dinitrobenzene. + – NO2 NH2 N2Cl (NH 4) 2S x OH H2O warm HNO2 Partial reduction HCl, 273 K NO2 NO2 NO2 NO2 (c) Sulphonation: Sulphonation of phenol with concentrated sulphuric acid produces o- and p-phenolsulphonic acids. OH OH Conc. H2SO4 373K SO3H + HO SO3H Chapter 4 OH 170 ORGANIC CHEMISTRY [VOL-II] At low temperature (293 K) o-isomer predominates but at high temperature (373 K) p-isomer is the major product. Further, when o-isomer heated to 373 K it gives thermodynamically more stable p-isomer. (d) Friedel-Crafts alkylation: Phenols, upon treatment with alkyl halides, in presence of anhydrous aluminium chloride produce ortho- and para-alkylphenols. Care must be taken to ensure that excess of aluminium chloride is used because phenol reacts with aluminium chloride to form chloroaluminium salt which is more reactive than phenol. + OH H O – – + AlCl3 OAlCl2 –HCl + AlCl3 Chloroaluminium salt (More reactive than phenol) The chloroaluminium salt reacts with alkyl halide yielding mainly ortho- and para-alkyl phenols in presence of the additional amount of aluminium chloride. – + OAlCl2 OH OH R + R X AlCl3 excess + R As in the case of benzene, alkylation of phenols can also be effected with alkenes in presence of aluminium chloride. For instance, m-cresol can be converted into thymol on treatment with propene. CH3 CH3 + CH3 CH CH2 AlCl3 excess OH OH Thymol (e) Friedel-Crafts acylation: Acylation of phenols to ortho- and para- hydroxyketones can be effected by treatment of phenol with acid chlorides or acid anhydrides in presence of a large excess of aluminium chloride. However, the best procedure to obtain such compounds is through the Fries migration of phenolic esters as discussed earlier. (vii) Gatterman synthesis: Gatterman formylation: It is an important synthetic procedure for preparing phenolic aldehydes. It involves treatment of phenol with a mixture of hydrogen cyanide and hydrogen chloride in presence of anhydrous aluminium chloride. OH HCN / HCl/ lCl3, 313K HO CHO p-Hydroxybenzaldehyde PHENOLS 171 Mechanism: It is an electrophilic substitution involving the attack of formaldiminium ion (I) formed from hydrogen cyanide and hydrogen chloride on aromatic ring, giving rise to an aldimine (II) which on hydrolysis with mineral acid affords the formyl derivative of phenol. – + H H + Cl Cl Cl + H C + N + H H C + NH H C NH – Cl Formaldiminium ion (I) H C NH Formaldimino chloride H N C Cl + AlCl3 H δ+ C N δ– AlCl3 Cl – + H N H C + AlCl4 H Formaldiminium ion (I) – + OH H + – AlCl3 O OAlCl2 –HCl + AlCl3 Chapter 4 H Chloroaluminium salt – + OAlCl2 O + OH + H C NH H CH NH CH NH II OH OH + H OH H2O H + O CH + + NH2 CH NH2 CH NH2 H + + –H +H + –H –NH3 H O CH OH OH OH + NH3 H + O CH CHO 172 ORGANIC CHEMISTRY [VOL-II] The formyl group goes to para-position with respect to phenolic group and if this position is blocked then it goes to ortho-position. (viii) Hauben-Hoesch reaction: It is an extension of Gatterman reaction and involves acylation of highlly reactive polyhydric phenols having hyroxyl groups in the meta-position with respect to each other. The reaction is carried out by treating these phenols with alkyl cyanide and hydrogen chloride in presence of anhydrous zinc chloride or aluminium choride. The ketimine produced is hydrolyzed with aqueous acid. COCH3 HO OH 2. H 3O OH HO 1. CH3CN/ HCl /AlCl3 + OH OH Phloroglucinol Phloroacetophenone (2, 4, 6-Trihydroxyacetophenone) Mechanism: The following mechanistic scheme, which is similar to that of Gatterman reaction, appears plausible: + CH3 C + AlCl3 N + H CH3 C + NH CH3 C NH CH3 H HO OH + C NH OH HO CH3 + C + NH OH OH Phloroglucinol + –H COCH3 HN OH HO + H3O C CH3 OH HO OH OH Phloroacetophenone Ketimine (ix) Carboxylation-Kolbe reaction (also called Kolbe-Schmidt reaction): Sodium salt of salicylic acid is obtained by passing carbon dioxide gas over heated sodium phenoxide (453–473 K) under pressure. This is also an example of electrophilic aromatic substitution reaction involving a very weak electrophile—carbon dioxide. PHENOLS 173 – + OH OH ONa – + COOH COONa + CO2 HCl –NaCl 353–473K 4–7 atm Sodium phenoxide Sodium salicylate Salicylic acid At lower temperatures the ortho isomer predominates whereas para isomer is obtained in excess at higher temperatures. Since in this reaction a—COOH group is directly introduced in the ring, it is also known as carboxylation. Mechanism: The most probable mechanistic pathway for the reaction may involve the electrophilic attack of carbon dioxide on activated ortho position of the phenoxide ion. – O OH O O C C + –H + +H – C OH – H + O O O COOH O + +H It may be noted that under the above conditions only half of the phenol is carboxylated whereas at lower temperature (393–413K) and under high pressure (5–7 atmospheres), phenol is completely converted into the product. This modification is referred to as Kolbe-Schmidt reaction. (x) Reimer-Tiemann reaction: The most successful method for the formylation of aromatic ring of phenols consists in treating phenols in alkaline solutions with chloroform at a temperature lower than the boiling point of the latter. This reaction, known as the Reimer-Tiemann reaction, usually results in the formation of ortho isomer as a major product which can be separated from the para isomer by steam distillation after acidification. O – OH CHO CHO + H3 O (Major) OH + CHCl3 – OH 340K + – O OH + H3 O CHO (Minor) CHO If instead of chloroform, carbon tetrachloride is used, salicylic acid is obtained as major product along with a small amount of p-hydroxybenzoic acid. Chapter 4 O 174 ORGANIC CHEMISTRY [VOL-II] OH OH OH COOH (i) NaOH, 340K (ii) H O + + CCl4 + 3 Salicylic acid (Major product) COOH p-Hydroxybenzoic acid (Minor product) Mechanism: The mechanism of this reaction involves electrophilic substitution on the phenoxide by dichlorocarbene (I) which in turn is produced by 1,1-ElcB reaction of chloroform. The resulting benzal derivative (II) undergoes alkaline hydrolysis to produce the corresponding aldehyde. – OH + H CCl3 Fast – – H2O + CCl3; Cl2C Slow Cl – + Cl CCl2 Dichlorocarbene (I) – O O O H CHCl2 + –H + +H – + CCl2 – CCl2 II O – H O Cl C Cl – O Cl C – –Cl O – OH CHO – –Cl –H2O H – – II H CHO + H3O + OH + OH The predominance of ortho isomer may be due to its greater stability resulting from intramolecular hydrogen bonding (chelation). Further, due to intramolecular hydrogen bonding, ortho-isomer having lower boiling point can be separated from para-isomer by steam distillation. H C O O N (xi) Coupling reaction: Ice cold solutions of phenols on treatment with arenediazonium salt solutions in weakly alkaline medium form brilliantly coloured compound called azo dyes. + – – N2Cl + O Benzenediazonium chloride Phenoxide pH 9 - 10 N N p-Hydroxyazobenzene (an orange dye) OH PHENOLS 175 (xii) Lederer-Manasse reaction: Phenols condense with aliphatic and aromatic aldehydes in presence of either an acid or a base as a catalyst. The process can be exemplified by the reaction of formaldehyde with phenol in presence of acid or alkali forming o- and p-hydroxybenzyl alcohols (o-hydroxymethylphenol (I) and p-hydroxymethyl phenol (II)) which are important starting materials for the manufacture of a phenol formaldehyde resin called Bakelite. OH OH CH2OH + CH2 Acid or base cold O + HO CH2OH II (Major) I ( Minor) But the reaction does not stop at this stage and the phenolic alcohols (I and II) condense with further amount of formaldehyde to form bishydroxymethyl phenols (III and IV) or with further amount of phenol to give all the possible dihydroxydiphenylmethanes (V, VI and VII). OH CH2OH 2CH2 O – + H or OH HOH2C CH2OH + Chapter 4 I + II OH CH2OH III AND IV HO CH2 OH V + OH I + II + HO CH2 O – + H or OH HO CH2 VI + HO HO CH2 VII Such condensations are repeated and finally a polymeric resinous mass called Bakelite is obtained. Bakelite is a thermosetting plastic and is very hard and rigid. 176 ORGANIC CHEMISTRY [VOL-II] CH2 CH2 CH2 CH2 HO OH HO CH2 III + IV + V + VI + VII CH2 CH2 HCHO, – + H or OH HO OH CH2 CH2 CH2 OH CH2 HO CH2 CH2 Phenol - formaldehyde polymer (Bakelite) Mechanism: This also involves an electrophilic attack of formaldehyde or its conjugate acid on phenoxide (alkaline medium) or phenol (acidic medium), respectively. Alkaline medium (dilute alkali) O H O – OH – O O CH2 H O CH2O O – CH2O – OH – H2O – CH2OH Acidic medium (dilute acid) + CH2 O + H CH2 + + OH CH2 OH + OH OH H + + CH2 OH OH CH2 + OH –H CH2 OH – PHENOLS 177 The mechanism of the polymerization in acidic and alkaline media may be depicted as under: Alkaline medium – O CH2 – OH O + H2 C O O + – CH2 O CH2 O CH2 O – H – OH O H2 C – etc. O – CH2 O HO CH2 OH + H HO Chapter 4 Acidic medium + OH2 CH2 –H2O + + HO HO H2 C CH2 + HO CH2 OH O CH2 + HO – H –H + +H + + H2 C etc. OH HO CH2 OH (xiii) Hydroxylation: Phenols, particularly polyhydric phenols, upon fusion with alkali form the product(s) having more hydroxyl groups attached to the aromatic ring. For instance, when quinol is heated rapidly with a large excess of sodium hydroxide, hydroxyquinol is formed together with other products. The reaction probably involves nucleophilic displacement of hydride by hydroxide ion. 178 ORGANIC CHEMISTRY [VOL-II] OH OH – OH H – + OH OH OH OH – –H OH OH Most convincing evidence for the above mechanism comes from the fact that hydrogen is evolved during the course of this reaction. (xiv) Oxidation: As the phenols are able to readily donate electrons(s) to various oxidizing agents, they are susceptible to oxidation. This is the reason why phenols turn pink or brown on exposure to air and light. The colour change may be attributed to the formation of quinones and phenoquinones. Different products are obtained using different reagents. Some examples are discussed below: (a) Oxidation with alkaline KMnO4 or stronger oxidising agents: Oxidation of phenols with stronger oxidising agents such as alkaline KMnO4 leads to the cleavage of the aromatic ring. However, if the —OH group is protected by alkylation or acylation, the alkyl side chain can be oxidised to give the corresponding hydroxy acids. – CH3 CH3 COO CH3COCl COOH + H 3O KMnO4 –HCl OH O COCH3 O COCH3 OH (b) Oxidation with weaker oxidising agents such as ferric chloride: Oxidation of phenols with ferric chloride leads to the formation of a phenoxyl radical which is resonance stabilized and undergoes radical coupling reaction at ortho-ortho, ortho-para, para-para positions, as shown below: O H + Fe O O +3 O H O + Fe+2 H H Radical coupling O O O H H + ortho-ortho coupling H ortho-para coupling O + O O H para-para coupling PHENOLS 179 (c) Oxidation with alkaline potassium persulphate: Monohydric phenols, upon treatment with potassium persulphate in alkaline solution, undergo oxidation yielding dihydric phenols. The —OH group enters preferentially at p-position if it is free otherwise it goes to o-position. This reaction is known as Elbs persulphate oxidation. K2S2O8 – OH OH OH OH Hydroquinone Phenol OH K2S2O8 – OH OH CH3 CH3 p-Cresol OH 4-Methylcatechol Mechanism H – – O O SO3 – SO3 O SO3 O SO3 + SO4 + – HO 2– O Chapter 4 O –H + H3O OH – – O (xv) Liebermann’s nitroso reaction: Phenol, upon treatment with sodium nitrite and conc. H2SO4, gives a deep green or blue colour which changes to red on dilution with water. When the resulting solution is made alkaline with sodium hydroxide the green or blue colour is restored. This reaction, known as Liebermann’s nitroso reaction is used as a test for the detection of phenol. OH O H O N O p-Nitrosophenol N Sodium salt (Blue or green) OH H2SO4 N OH Quinone monoxime – + O H + –H + +H NaNO 2 / HCl ONa NaOH O N OH Phenol - indophenol ( Red ) (xvi) Formation of phthaleins or fluoresceins: On heating with phthalic anhydride, phenols (in the ratio 1 : 2) in presence of an acid catalyst such as anhydrous ZnCl2 or AlCl3 or conc. H2SO4 afford phthalein or fluorescein dyes which give characteristic colour in alkaline medium. Whereas monohydric phenols yield phthaleins, polyhydric phenols give fluoresceins. 180 ORGANIC CHEMISTRY [VOL-II] OH HO O H C O OH + C C H2SO4 –H2O H O C OH O O Phenol (Monohydric) Phthalic anhydride Phenolphthalein (pink in alkaline medium) OH HO O O OH C C O H2SO4 –H2O + 2 C O O C OH Resorcinol (Dihydric) O Fluorescein (yellow-green fluorescence in alkaline medium) Phthalic anhydride Phenolphthalein is used as an indicator in acid-base titrations. It is colourless in acidic medium and turns pink in alkaline medium. (xvii) Reduction: Phenols on heating with zinc dust, form aromatic hydrocarbons. OH + Zn Δ + ZnO However, catalytic reduction gives cycloalkanols involving saturation of the ring. OH OH + 3Zn 4.8 Ni 433 K DISTINCTIONS BETWEEN ALCOHOLS AND PHENOLS (i) Litmus test: Phenols turn blue litmus red but alcohols do not. (ii) Coupling reaction: Phenols react with diazonium salts at pH 9-10 to form yellow or orange azodyes but alcohols do not. PHENOLS 181 (iii) FeCl3 test: Phenols react with neutral FeCl3 to give blue, violet or green colour but alcohols do not. (iv) Br 2/H 2O test: Phenols give a white precipitate of polybromophenols (e.g., 2, 4, 6-tribromophenol in case of phenol) but alcohols do not. A BRIEF REVIEW Phenols are obtained by: (i) treating halobenzenes with NaOH under suitable conditions (Dow process). (ii) fusion of sodium salts of aromatic sulphonic acids with NaOH at 573–623 K followed by acidification with dil. H2SO4. (iii) hydrolysis of benzene diazonium salts with boiling dil. H2SO4. (iv) phenol itself is obtained from cumene by catalytic aerial oxidation. Phenols are stronger acids than alcohols. The phenoxide ion is resonance stabilized while alkoxide ion is not. Effect of substituents on acid strength of phenols (i) Electron-donating groups decrease the acid strength of phenols. Thus the order of acid strength is phenol > m-cresol > p-cresol > o-cresol. (ii) Electron-withdrawing groups increase the acid strength of phenols. Thus the order of acid strength is p-nitrophenol > o-nitrophenol > m-nitrophenol > phenol Effect of position of substituents. The increase or decrease in acid strength of phenols is more pronounced at o- and p-positions than at m-position because a group present at m-position can not enter into resonance with the phenoxide ion. m-Methoxyphenol and m-aminophenol are, however, more acidic than phenol due to –I-effect of the —OCH3 or the —NH2 group, i.e, m-methoxyphenol > phenol > o-methoxyphenol > p-methoxyphenol. Sodium or potassium phenoxide reacts with alkyl halides to form phenolic ethers. The reaction is called Williamson’s synthesis Allyl phenyl ethers when heated to 475 K, undergo Claisen rearrangement to form o-allylphenols. If both the ortho positions are occupied, the allyl group migrates to p-positions. This reaction, occuring by a concerted mechanism through a six-membered cyclic transition state, is also called as Sigmatropic rearrangement. Phenols react with acid chlorides or anhydrides in presence of pyridine to form esters. With acetyl chloride or acetic anhydride, phenol gives phenyl acetate. It is called acylation. Benzoylation of phenol with benzoyl chloride in presence of NaOH gives phenyl benzoate (Schotten-Baumann reaction). The acetyl group migrates to o- and p-positions to form a mixture of o-hydroxyacetophenone and p-hydroxyacetophenone when phenyl acetate is heated in presence of anhyd. AlCl3. Under similar conditions, phenly benzoate gives a mixture of o- and p-hydroxybenzophenone. This reaction is called Fries rearrangement and usually occurs in two steps. Phenols but not alcohols on distillation with zinc dust give the corresponding aromatic hydrocarbons. Due to the presence of strongly activating OH group, phenols readily undergo electrophilic substitution reactions. (i) With conc. HNO3 in presence of conc. H2SO4, phenol gives picric acid. But the yield is poor. (ii) With dil. HNO3 at 293 K, phenol gives a mixture of o-nitrophenol (major) and p-nitrophenol (minor). Chapter 4 182 ORGANIC CHEMISTRY [VOL-II] (iii) With conc. H2SO4 at 288–293K, phenol gives o-phenolsulphonic acid as the major product, but at 373 K it gives p-phenolsulphonic acid as the major product. (iv) With Br2 in CS2 at 273 K, a mixture of p-bromophenol (major) and o-bromophenol (minor) is obtained. (v) With Br2 water phenol gives 2,4,6-tribromophenol. (vi) With alkyl halides in presence of anhyd. AlCl3, phenol give a mixture of o-alkylphenol (minor) and p-alkylphenol (major). (vii) Sodium phenoxide when heated with CO2 at 400K under a pressure of 4–7 atmospheres followed by acidification gives salicylic acid (Kolbe’s reaction). Since by this reaction, a carboxyl group is directly introduced into the aromatic ring, this reaction is also called carboxylation. (viii) Phenol mainly gives p-hydroxybenzaldehyde when treated with a mixture of HCN + HCl in presence of anhyd. AlCl3. This reaction is called Gattermann formylation because a formyl group is directly introduced into the aromatic ring. (ix) Houben-Hoesch reaction is an extension of Gattermann formylation and involves acylation of highly reactive polyhydric phenols having two or three hydroxyl groups at m-position to each other. The reaction is usually carried out by passing HCl gas through a cold ethereal solution of a polyhydric phenol and acetonitrile in presence of anhyd. AlCl3 or ZnCl2 when the ketimine hydrochloride gets precipitated. This upon subseqent hydrolysis with boiling water gives the corresponding phenolic ketone. Under these conditions, phloroglucinol gives phloroacetophenone. (x) Phenol reacts with CHCl3 in presence of NaOH at 340 K to form salicylaldehyde as the major product alongwith a small amount of p-hydroxyaldehyde. The reaction involves electrophilic attack of dichlorocarbene on phenoxide ion and is called Reimer-Tiemann reaction. If CCl4 is used in place of CHCl3, salicylic acid is the main product. (xi) When phenol is treated with formaldehyde in presence of an acid or a base, it initially gives a mixture of o-and p-hydroxymethylphenols. This reaction is called Lederer-Manasse reaction. The phenolic alcohols thus produced subsequently undergo polymerization to form a highly crosslinked threedimensional polymer called Bakelite. (xii) Phenol condenses with phthalic anhydride in presence of conc. H2SO4 to form phenolphthalein which is used as an indicator in acid-alkali titrations. (xiii) Phenol reacts with benzenediazonium salt in presence of weakly alkaline solution (pH 9–10) at 273–278 K to form brilliant coloured compounds called azo dyes. Distinction of phenols from alcohols. Phenols (i) turn blue litmus red (ii) give blue or violet colour with neutral FeCl3 (iii) produce white ppt. with bromine water and (iv) form yellow or orange coloured azo dyes with diazonium salts. Alcohols do not respond to these tests. SOLVED PROBLEMS Q.1. Explain why alochols react with organic acids to form esters but phenols do not. Ans. Nucleophilic attack of alcohols occurs on the protonated carbonyl group of carboxylic acids during formation of esters. + OH O OH + R OH + R C OH R O C H OH R + –H2O, –H R O C R Protonated acid But phenols, are less nucleophilic than alcohols because of resonance which deplete the electron density on oxygen. Due to this they fail to attack the protonated carbonyl group of acids to form esters. PHENOLS 183 H H + O + O H + O H O OH – – – Resonating forms of phenol Q.2. Explain why phenols do not undergo substitution of the —OH group in contrast to alcohols? Ans. The C—O bond in alcohols has single bond character and hence can be cleaved by a nucleophile. But the C—O bond in phenols has some double bond character due to resonance and hence cannot be easily cleaved by a nucleophile. Q.3. Explain why the compound I is more acidic than? OH OH CH3 H3C CH3 NO2 NO2 I II Ans. In phenol II, the nitro group is flanked by two groups which push the nitro group out of the plane of the benzene ring. As a result of this steric hindrance, the electron-withdrawing resonance effect of the nitro group will be reduced. However, in phenol I, no such steric inhibition of resonance occurs. In other words, the phenoxide ion obtained from I is better stabilized by the nitro group than the phenoxide ion derived from phenol II. Thus, phenol I is more acidic than II. Q.4. Alcohols react with halogen acids or phosphorus halides to form haloalkanes but phenols do not form halobenzenes. Explain. Ans. The C—O bond in phenols has some double bond character due to resonance and hence cannot be – easily cleaved by X ions in presence of halogen acids or phosphorus halides to form halobenzenes. – In contrast, the C—O bond in alcohols is a single bond and hence can be easily cleaved by X ions in presence of halogen acids or phosphorus halids to form haloalkanes. Q.5. Explain why dipole moments of phenol (1.7D) and methanol (1.6D) are in opposite directions. Ans. Delocalization of electron-density from O to the benzene ring makes the O of phenols to be the positive end of the molecular dipole. In alcohols, the strongly electron-withdrawing oxygen is the negative end of the dipole. Thus, the two dipoles act in opposite directions. Q.6. Account for the fact that phenols are much more stable than enols. Ans. Both phenols and enols can be considered as tautomers of the respective keto forms. The large resonance energy of C==O usualy makes the keto-tautomer much more stable than the enol form. However the large resonance energy of the aromatic ring makes phenol more stable than the keto form. O OH O H C H o-Keto form H H C Enol form OH H C C O Keto form p-Keto form Q.7. How do you account for the fact that unlike phenol, 2,4-dinitrophenol and 2,4,6-trinitrophenol are soluble in aqueous sodium carbonate solution? Chapter 4 H3C 184 ORGANIC CHEMISTRY [VOL-II] Ans. Due to strong electron-withdrawing nature of NO2 groups both 2,4-dinitrophenol and 2,4,6-trinitrophenol are more acidic than carbonic acid and hence decompose Na2CO3 solution to form the corresponding sodium salts with the evolution of CO2. Q.8. Consider the following reaction sequence for the synthesis of benzaldehyde. C6H6 + CO + HCl AlCl3, 100 atm C6H5CHO + HCl If we use DCl in place of HCl, shall we get C6H5CHO or C6H5CDO? Give reason for your answer. – Ans. C + O + HCl + AlCl3 + HC – O + AlCl4 ⊕ If instead of HCl, DCl is used, then D — C == O will be formed which will attack the benzene ring to form deuterated benzaldehyde. C6H6 + CO + DCl AlCl3, 100 atm C6H5CDO + HCl Q.9. Out of o- and p-nitrophenols which one has higher boiling point and why? Ans. o-Nitrophenol exists as discrete molecules due to intramolecular H-bonding while p-nitrophenol exists as associated molecules due to intermolecular H-bonding. Therefore, p-nitrophenol has higher boiling point than o-nitrophenol. Q.10. How will you distinguish between the following pairs?: (i) Phenol and benzaldehyde (ii) Phenol and benzoic acid (iii) Phenol and ethyl alcohol Ans. (i) Phenol on heating with phthalic anhydride and a few drops of conc. H2SO4 gives phenolphthalein which gives pink colour with NaOH while benzaldehyde fails to give this test. (ii) Benzoic acid gives brisk effervescence with aqueous solution of NaHCO3 while phenol does not give this test, C6H5COOH + NaHCO3 → C6H5COONa + CΟ2↑ + Η2Ο (iii) Phenol gives purple colour with neutral FeCl3 solution whereas ethyl alcohol does not respond to this test. Q.11. m-tert-Butylphenol reacts with chlorine forming a trichloroderivative but with iodine it forms only monoiodo derivative. Explain. Ans. Steric hindrance by bulky tert-butyl group does not allow the large sized iodine atom to attack at positions ortho to it. As a result, I attacks only at o-position with respect to OH group to give monoiodo derivative. In contrast Cl atom is much smaller and hence attacks at all the o- and p-positions with respect to OH group to form trichloro derivative. Q.12. The boiling point of toluene is 384 K while that of phenol is 455 K. Explain Ans. More energy is required to break intermolecular H-bonds existing in phenol compared to weak van der Waals forces of attraction existing in toluene. Q.13. 2, 6-Di-tert-butylphenol is a much weaker acid than phenol. Why? Ans. Bulky o-tert-butyl groups prevent solvation of the corresponding phenoxide anion thus making it less stable than phenoxide ion. Q.14. What are the principal ions in solution when the following are mixed? (i) Sodium ethoxide and phenol (ii) Sodium phenoxide and ethanol. Ans. (i) C2H5ONa + C6H5OH ⎯→ C2H5OH + C6H5ONa. Phenol being a stronger acid than alcohol displaces ethanol from sodium ethoxide. Thus ethanol and sodium phenoxide are formed. (ii) Nothing will happen. Q.15. Arrange the following in increasing order of solubility in water: C2H5OH, C6H5CH2OH, C6H5OH. PHENOLS 185 Ans. Solubility decreases as the length of the carbon chain or the size of the hydrocarbon part increases. Thus, solubility increases in the order: C6H5CH2OH < C6H5OH < C2H5OH. Q.16. Treatment of phenols with alkyl halides in presence of KOH results in the formation of only O-alkylated produts by SN 2 process. But 2, 6-di-tert-butylphenol give a mixture of C-alkylated and O-alkylated products. Further, the amount of C-alkylated product increases with the increase in the size of alkyl group of alkyl halides. Explain. Ans. In presence of KOH, SN 2 attack of phenoxide ion occurs on alkyl halide to form alkyl aryl ethers. – – Ar O + R X ArO R + X However, SN 2 attack of 2,6-di-tert-butylphenoxide ion on alkyl halide is sterically hindered due to the presence of bulky tert-butyl groups at o-positions. The negative charge is also available at p-position due to resonance. Since this position is sterically unhindered, preferential nucleophilic attack occurs at this position to give C-alkylated product as the major product. O Sterically hindered R – O O – O X R – –X O X – –X + –H – O-Alkylated product (not formed) H R R C-Alkylated product Further, as expected, as the size of the alkyl group in alkyl halide increases, steric hindrance for Oalkylation increases. Therefore, the amount of O-alkylated derivative decreases while that of C-alkylated product increases. Q.17. Houben-Hoesch reaction occurs with polyhydric phenols having two or three hydroxyl groups at m-position with respect to each other but not with phenols having hydroxyl group at o- and p-positions. Why? Ans. Monyhydric phenols react with alkyl cyanide and HCl in presence of anhydrous AlCl3 (Houben-Hoesch reaction) to form mainly imido esters. Ar OH + HCl + R C N R Anhydrous AlCl3 + C Ar O – NH2Cl Imido ester However, when two or three OH groups are present at m-position with respect to each other, the reactivity of the nuclear positions, o- and p- to the hydroxyl group increases to such an extent that nuclear acylation occurs preferentially. The reactivity of the nuclear position does not increase significantly when the two OH groups are at o- and p-positions and hence the reaction does not occur with phenols having two hydroxyl groups at o- and p-positions with respect to each other. UNSOLVED PROBLEMS 1. What are phenols? Describe three methods by which benzene can be converted into phenol. 2. Why are phenols more acidic than alcohols? Discuss the effect of substituents on the acid strength of phenols. Chapter 4 R 186 ORGANIC CHEMISTRY [VOL-II] 3. Compound (A), C7H8O is insoluble in aqueous sodium bicarbonate but dissolves in aqueous sodium hydroxide and gives characteristic colour with aq. ferric chloride. When treated with bromine (A) forms a compound (B), C7H5—OBr2. (i) Give the structural formulae for (A) and (B). (ii) What would be the structure of compound (A) if it neither dissolves in aqueous sodium hydroxide nor gives a characteristic colour with ferric chloride solution? 4. Write IUPAC names of the following compounds: OH H3C OH OH CHO ( i) OH CHO ( ii) (iii) H3C NO2 Br (iv) COOCH3 NO2 CH3 Cl 5. What is cumene? How can it be prepared and how can it be converted into phenol? 6. Describe Dow’s process for the manufacture of phenol. Comment upon its mechanism. 7. Explain the following: (a) Phenol has higher boiling point than toluene. (b) Phenol is more soluble in water than toluene. (c) o-Nitrophenol has lower melting point and decreased water solubility as compared to its m- and p-isomers. (d) p-Nitrophenol is a stronger acid than phenol. 8. Discuss the mechanism of the following: (a) Fries rearrangement (b) Claisen rearrangement (c) Gatterman synthesis (d) Hauben-Hoesch reaction (e) Reimer-Tiemann reaction (f ) Lederer-Manasse reaction 9. How does phenol react with the following reagents? (a) Zinc dust/Δ (b) Br2/H2O (c) HNO3 (d) Conc. H2SO4 (e) HNO2 (f ) H2/Ni 10. Identify compounds A, B, C and D in the following sequence of reactions: o-Nitrophenol (CH3)2SO4 NaOH A Zn, HCl B NaNO2, HCl 273K C C2H5OH D 11. How would you distinguish between o- and p-hydroxybenzaldehydes on the basis of infrared spectral studies? 12. Compare the acid strengths of (a) p-Chlorophenol and p-nitrophenol (b) 2,4-Dinitrophenol and 2,4,6-trinitrophenol (c) o-Aminophenol and p-aminophenol 13. Arrange, giving appropriate reasoning, the following sets in order of increasing acid strength: (a) Benzenesulphonic acid, benzyl alcohol, phenol, benzoic acid. (b) Phenol, o-nitrophenol, p-nitrophenol 2,4-dinitrophenol. 2,4,6-trinitrophenol. PHENOLS 187 (c) Phenol, m-anisole, p-chlorophenol, quinol, p-aminophenol, p-nitrophenol. (d) m-Nitrophenol, m-bromophenol, m-cresol and phenol. 14. Why does phenol turn pink on exposure to air and sunlight? Hint. Phenol turns pink on exposure to air and light due to slow oxidation to produce quinone. OH + O2 O O + H2O Quinone then combines with phenol through hydrogen bonding to form brilliant red adduct known as phenoquinone. OH + O O + HO OH O O HO 15. Explain, why phenols couple more readily in slightly basic than in acidic solution. 16. Introduction of nitro group in aromatic ring increase the acidic character of phenol while introduction of methyl group decreases its acid strength. Explain. 17. Complete the following reaction and name the products and the reaction involved. ONa + CO2 398K, 1 – 7 atom + H 18. How will you synthesize butyl phenoxyethanoate (A) using phenol, acetic acid and any other chemical you wish? OCH2COOCH2CH2CH2CH3 (A) 19. (i) With the help of resonance contributing structures, show how the presence of a nitro group in the ortho and para positions stabilizes the phenolate anion while that in the meta position it does not? (ii) Why does p-nitrophenol not form intramolecular hydrogen bond? 20. Predict the products in the following transformations: (i) p-Cresol 1. NaOH 2. CO2, 373K, Pressure 3. H (iii) p-Chlorophenol HNO3, H2O (ii) p-Cresol + 2Br2 (iv) p-Bromophenol + (CH3)2SO4 – OH H2O Chapter 4 Phenoquinone (Brilliant red compound) 188 ORGANIC CHEMISTRY [VOL-II] – OH (v) C6H5OH + ClCH2COOC2H 5 21. Predict the products(s) and sketch a plausible mechanism for each of the following reactions: OH CHO K2S2O8 (i) (ii) NaOH + H2O2 OH Cl OH (iii) OH + (CH3CO)2O + H (iv) + C6H5COCl – OH OH (v) – CHCl3, OH 22. Sketch the mechanism for the following transformation: CH(CH3)2 OH 1. O2, 368–408K 2. Dilute H2SO4 H3 C C + O H3C ❑❑❑