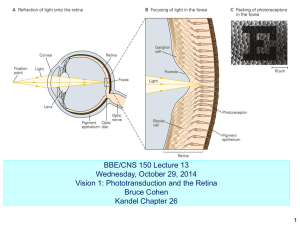
Introduction to CGMP
advertisement

FDA cGMP Training Program cGMP in the USA Nicholas Buhay Deputy Director Division of Manufacturing & Product Quality Office of Compliance, CDER, FDA Introduction to Drug Current Good Manufacturing Practice Of US FDA An Outline • • • • • • Legal bases for CGMP CGMP legal principles CGMP Implementation Tools CGMP Resources Overview of CGMP Requirements Integrity of Records and Data FD&C Act; 501(a)(2)(B) “A drug shall be deemed adulterated if: ... the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice ...” more... FD&C Act; 501(a)(2)(B) “to assure that such drug meets the requirements of this Act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.” CGMP legal principles • Quality built into product – By “taking care” in making medicine – Can’t ‘test’ into product the quality • Without/Inadequate CGMP – Product(s) adulterated(defects need not be shown) – Firm and its management are responsible CGMP legal principles • Non-compliance = eventual problems – Superpotency/subpotency – Contamination – Misbranding – Bioavailability – Safety and efficacy CGMP Legal Principles • Scope – Ingredients (APIs + excipients) – Finished dosage forms administered to humans/animals » OTC, Rx products » Biologics, veterinary drugs » Drugs undergoing study(IND, etc) – Manufacturers, test laboratories, packagers(including pharmacies) CGMP legal principles • Excluded from the CGMP requirement – Positron emission tomography, per FDAMA (own CGMP to be developed) – Drug products compounded per Section 503 Pharmacy Compounding (FDAMA) CGMP Legal Principles • Current = dynamic – Standards evolve over time • Good practices – Minimal standards – Not “best practices” » Unless “best” is, in fact, current minimal CGMP Legal Principles • Feasible and valuable – No threshold for “percentage” in practice » Doesn’t have to be “predominant” – Enforceable even if nobody is doing it » Stronger case if someone is doing it The CGMP Regulation • CGMP for Finished Pharmaceuticals 21 CFR 210, 211 – First issued: June 1963 – Today’s version: September 1978 – Scope » Dosage forms for human/vet/biologics » OTC, Rx, IND, NDA, Medical Gases » Not: pharmacies, ingredients, non-clinical research, etc The CGMP Regulation • CGMP for Finished Pharmaceuticals 21 CFR 210, 211 – Substantive » Force and effect of law – Constitute major part of (not entire) CGMP more... The CGMP Regulation • CGMP for Finished Pharmaceuticals 21 CFR 210, 211 – Establish “what to” do, not “how to” do » Minimal standards » Maximum flexibility » Specific enough to address problems • e.g., Penicillin contamination control » Technology neutral » Scalable CGMP Implementation Tools • Compliance Policy Guides – Specific actions we do related to CGMP – Examples: » Sub Chapter 410 Bulk Drugs • The regulations for finished pharmaceuticals will be applied as guidelines for bulk drugs » Sub Chapter 420 Compendial (USP)/Test Requirements Ex:USP not required for release test » Other Sub Chapters • • • • Labeling and Repackaging Stability/Expiration Process Validation Etc CGMP Implementation Tools • CGMP Guidance Documents – Principles: » Not requirements » Agency “current thinking” » Detailed, technical » Expression of “How to” meet “what to” do (requirements) – Shape industry behavior » offers routes to efficiency in meeting CGMP requirement, evaluation of compliance CGMP Implementation Tools • CGMP Guidance Documents (Examples) – General Principles of Process Validation – Compressed Medical Gases – Sterile Drug Products Produced by Aseptic Processing – Guideline on the Preparation of Investigational New Drug Products more... CGMP Implementation Tools • CGMP Guidance Documents – Investigating Out of Specification Test Results for Pharmaceutical Production – Manufacturing, Processing or Holding of Active Pharmaceutical Ingredients CGMP Implementation Tools • CGMP Compliance Programs – Instructions to FDA inspectors – Drug Manufacturing Inspections Program » Systems-based assessment of site – Preapproval Inspection Program » Points to inspect » Laboratory support » Regulatory approaches CGMP Implementation Tools • CGMP Guides to Inspection of…. – Help field investigators apply CGMP » Uncover need for CGMP changes » Specific to topics (e.g., cleaning validation) CGMP Resources • Internet WWW site by DMPQ – http://www.fda.gov/cder/dmpq » CGMP regulations and ongoing changes » Preamble to the CGMP regulation » Division subject contacts » Medical gases » Active pharmaceutical ingredients » Human Drug CGMP Notes/Policy » etc. Overview of CGMP requirements in the regulation • CGMP Regulations – 21 CFR 210 » Status of the regulations » Applicability of the regulations » Definitions • • • • • • Batch Lot In-process material Quality control unit Representative sample etc Overview of CGMP requirements in the regulation • CGMP Regulations – 21 CFR 211 » Subpart A General Provisions » Subpart B Organization an Personnel » Subpart C Buildings and Facilities » Subpart D Equipment » Subpart E Control of Cmpnts/Cntr/Closures » Subpart F Production and Process Controls » Subpart G Packaging and Labeling Controls more... Overview of CGMP requirements in the regulation • CGMP Regulations – 21 CFR 211 » Subpart A General Provisions • this is minimum CGMP Overview of CGMP requirements in the regulation • CGMP Regulations – 21 CFR 211 » Subpart B Organization and Personnel • There shall be a quality control unit • quality control unit responsibility to approve/reject Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart C Buildings and Facilities • buildings shall be….suitable • operations to be in specifically defined areas….separate…. Or such other control systems for ….operations as are necessary to prevent contamination or mix-ups…. (see list, includes aseptic processing) • “separate” facilities for penicillin • building….shall be….clean and sanitary Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart D Equipment • surfaces ….shall not be reactive, additive, or absorptive • Equipment….shall be cleaned, maintained and sanitized…. Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart E Control of Components, Containers and Closures • containers and closures ….handled in a manner to prevent contamination. • Testing or examination of c/c/c’s • test to identify each component • tests on components for conformance with specs • test c/c/c’s microscopically, for adulterants, microscopically Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart F Production and Process Controls • written procedures for production and process control • formulated not less than 100 % • portions of components identified, examined by a 2nd person before dispensed for use in manufacture • sampling and testing of in-process materials and products, some specified • time limits • reprocessing allowed, but controlled Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart G Packaging and Labeling Controls • examination, approval of labels, labeling • strict control over labeling issue, and return to stock • written procedures, physical separation of labeling operations • examination of materials before use • inspection of facilities immediately before • tamper resistant packaging (for OTC products) • expiration dating Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart H Holding and Distribution » Subpart I Laboratory Controls » Subpart J Records and Reports » Subpart K Returned and Salvaged Drug Products Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart H Holding and Distribution • quarantine before release • store under appropriate conditions Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart I Laboratory Controls • establish specs, standards, sampling plans, test procedures • calibration, of laboratory equipment • test each batch of drug product • adequate acceptance criteria • validate test methods • conduct stability program more.... Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart I Laboratory Controls • Special tests – sterility and pyrogenicity – ophthalmic ointments for foreign/abrasive particles – controlled release products for rate of release • keep reserve samples • test non-penicillin products for penicillin when reasonable possibility of exposure to presence of penicillin Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart J Records and Reports • keep records, make available for inspection • conduct annual review of each drug product for changes to specs, control procedures • keep equipment cleaning and use log • keep component, container, closure and labeling records more.... Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart J Records and Reports • have SOP for master production and control record, maintain record • use batch production and control records for manufacture, keep records • records to be reviewed/approved by qual control unit • complete data derived from all tests necessary to assure compliance more.... Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart J Records and Reports • distribution records, with lot numbers(except medical gases) • complaint files Problem – Drug Regulatory Program depends heavily on the reliability (i.e. truthfulness, completeness and accuracy) of data & information in records – Applications for approval [AIP] – Manufacturing Controls documentation [non-AIP] – Historical experience with broad scale unreliability of data in records or in conduct related to records Data and records that are not acceptable or are misleading What are some characteristics of data that lack integrity? » Untrue, made up, false, no source in an event » Omission of significant data from the submission that is determined to be material to the review process. Data that is not submitted, but should have been » Inaccurate (e.g. First data failed specs, retest data passes specs, no lab investigation, but retest data is submitted to the application.) Records Must be True • All data and information in records submitted to FDA & supporting documents in the possession of the applicant are accurate & true representations of – Actual tests performed & the test results – Actual manufacturing & quality control steps & procedures associated with the development and manufacture of the submission batch (clinical/pilot or biobatch) – any other actions and conditions associated with the application Wrongful Acts Any act or conduct that subverts the integrity of the review process, including, but not limited to the following: – submitting fraudulent applications – offering or promising a bribe or illegal gratuities – making an untrue statement of a material fact (e.g. false statement, a misstatement or an omission of a fact) – submitting unreliable data which results from system-wide or firm-wide behavior Wrongful Acts (continued) An untrue statement of material fact is a false statement, a misstatement or an omission of a fact that is important in the review process. System-wide incompetence is also a wrongful act When an untrue statement of material fact or system-wide incompetence is found, several steps are required to the invoke the AIP including: – Documentation of a pattern or practice of wrongful acts. – Ensuring that the untrue statements are material facts. Pattern or Practice Pattern- More than one instance of errors or acts involving the subject matter important to the evaluation of an application Practice- An act or process of doing something affecting subject matter important to the evaluation of an application A practice can be one or more acts or processes. A pattern or practice can occur in one or more applications. If submitted to an Application The AIP procedures broadly define the term, “application” to include, but not be limited to, any application, amendment, supplement or other submission made by an applicant. “Submitted” is an understandable term and includes documents received by the review branch. Wrongful acts also include omissions of data and/or information that should have been submitted to an application. Food, Drug, and Cosmetic Act Section 505(e) (excerpt below) Numbered Part 5 – The Secretary shall, after due notice and opportunity for hearing to the applicant,withdraw approval of an application with respect to any drug under this section, if the Secretary finds…. – (5) that the application contains any untrue statement of a material fact TO INVOKE AIP Documentation of a pattern or practice of wrongful conduct that raises significant questions about the reliability of data submitted to an application - wrongful acts - pattern or practice - unreliable data Restore FDA’s Confidence in Data??? Cooperation with investigators Identification of involved individuals Credible internal review & actions Problem analysis/identify all instances of wrongful acts Use of impartial auditor/Outside consultant Audit Plan, audits, audit reports Other measures as FDA deems appropriate Restore FDA’s Confidence in Data Corrective Action Operating Plan: Analysis of audit findings Implementation of auditor recommendations Actions taken to correct fraud/wrongful acts, e.g. Withdraw applications & recall products Timetable Identification of persons assigned to complete and verify corrective actions Comprehensive ethics program Procedures for monitoring effectiveness of the plan Training in the requirements of the Act and 18 USC 1001 Corrective Actions Plan Evaluation Monitor applicant’s actions/inquiries during internal review Inspection to assess actions taken by applicant to determine if Internal Review performed adequately Corrective Action Operating Plan implemented adequately Submit recommendation to CDER to remove site from the policy Expect a long time to pass before restoration Overview of CGMP requirements • CGMP Regulations – 21 CFR 211 » Subpart K Returned and Salvaged Drug Products • if conditions cast doubt returned product shall be destroyed unless proved ok by test, examination, investigation • salvage only if evidence from tests and inspection show all standards met Input for CGMP Changes • Establishment inspections – Industry changes/problems • • • • • Defect reports/complaints/recalls Litigation Agency application reviews Trade/scientific literature Citizen petitions Management of CGMP Regulatory Program • FDA/CDER – OC/Division of Manufacturing and Product Quality – maintenance of the regulation – definitive interpretation – manage guidance development – develop, operate, evaluate programs – train FDA/outreach to industry We Have Discussed • • • • • • Legal bases for CGMP CGMP legal principles CGMP Implementation Tools CGMP Resources Overview of CGMP requirements Integrity of Records and Data Nicholas Buhay Deputy Director Division of Manufacturing and Product Quality, HFD-320 Center for Drug Evaluation and Research Phone: 301-827-8940 Fax: 301-827-8907 E-mail: buhay@cder.fda.gov Montrose Metro Centre II Room 438 11919 Rockville Pike Rockville, MD 20852 Return to Main Menu