ALZHEIMER DISEASE
advertisement

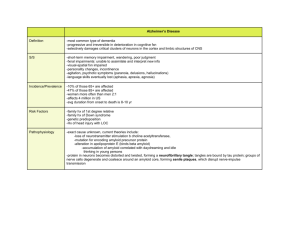
ALZHEIMER’S DISEASE Erin Dancey Overview Alzheimer’s is the most common cause of dementia in adult life and is associated with the selective damage of brain regions and neural circuits critical for memory and cognition The pathogenesis of this disease is complex, and involves many molecular, cellular, and physiological pathologies The neurons in the neocortex, hippocampus, amygdala, and the basal forebrain cholinergic system are the most affected brain regions Amyloid Plaque Formation Alzheimer’s patients show numerous plaques which are composed of 4 kD Amyloid-beta (A-beta) peptides, which are derived from beta amyloid precursor proteins (APPs) APP is a membrane associated glycoprotein of 110-135 kDa that is proposed to normally behave in the brain as a cell surface signaling molecule A-beta peptides are generated in the endosomal compartment and in the endoplasmic reticulum or golgi complex by endoproteolytic cleavage of APP by Beta, alpha, and gamma secretases Presenilins Presenilin 1 (PS1) and presenilin 2 (PS2) are highly homologous 43-50 kD proteins with eight transmembrane domains Presenilin’s make crucial contributions to neurodegeneration in AD Presenilin’s are crucial components of the enzymes that work to cleave APP, and mutations in presenilins cause the production of A-beta42 and A-beta43 peptides (insoluble forms of Abeta) The production of A-beta production by processing of APP Amyloid Plaque formation About 90% of the secreted A-beta peptides formed from processing of APP are A-beta40, a soluble form of the peptide About 10% of secreted A-beta peptides are Abeta42 and A-beta43 A-beta42 and A-beta43 are highly fibrillogenic, readily aggregated, and neurotoxic Amyloid Plaque formation Alignment of several strands of A-beta show that A-beta42 and A-beta43 preferentially form networks of salt linkages and strong hydrogen bonds between ionized side chains of opposite charge which thus form the observed plaques Alignment of the sequence of the 42 residue peptide of the plaque of Alzheimer’s disease Schematic representation of a parallel beta sheet in an amyloid fibril Micrographs of amyloid fibrils Micrograph of amyloid fibres Neurofibrillary pathology Intracellularly, alzheimer’s patients show neurofibrillary pathology Affected neurons accumulate tau and ubiquitin immunoreactivities within neurofibrillary tangles, in cell bodies and dendrites, and in dystrophic neuritis Plasmin In the brain, plasminogen and its proteolytic fragment are abundant in the hippocampus It has been hypothesized that brains of patients with AD may have lower levels of plasmin The higher production of amyloid peptide together with less efficient degradation would to A-beta accumulation and aggregation Early Onset Alzheimer’s Most cases of early onset AD are familial autosomal dominant disorders caused by mutations in APP, PS1, and PS2 Various substitutions have been studied and they have found various mutations that cause the individuals to secrete a higher fraction of A-beta42 and/or Abeta43 peptides Late Onset Alzheimer’s In late onset alzheimer’s, there are no specific gene mutations that are associated with the inheritance of the disease However, specific alleles of apoliprotein E4 (apoE) and alpha2 macroglobulin are associated with increased risk of alzheimers Amyloid Hypothesis The trigger for alzheimer’s disease is the A-beta peptide, and the accumulation of this peptide in the form of plaques is the initiating molecular event The plaques trigger an inflammatory response, neuronal cell death, and gradual cognitive decline The rest of the disease process, including formation of neurofibrillary tangles containing tau protein, is caused by an imbalance between A-beta production and A-beta clearance The sequence of pathogenic events leading to AD proposed by the amyloid hypothesis Postulated evolution of structural abnormalities and evidence of Abeta deposits in the hippocampus Support for the Amyloid Hypothesis The A-beta peptide is the primary component of the necrotic brain tissue Mutations in the gene encoding the tau proteins cause frontotemporal dementia with parkinsonism However, parkinsonism is characterized by severe deposition of tau in neurofibril tangles in the brain, but there is no deposition of amyloid Support for the Amyloid Hypothesis Growing evidence indicates that genetic variability in A-beta catabolism and clearance may contribute to the risk of late onset of AD Problems with the amyloid hypothesis The number of amyloid deposits in the brain do not correlate well with the degree of cognitive impairment that the patient experienced in life. In some cases, individuals without symptoms of AD have many cortical A-beta deposits. However, in these cases, these are diffuse amyloid plaques that are not associated with surrounding necrotic and glial pathology The amyloid hypothesis remains controversial because a specific neurotoxic species of A-beta and its effects on neuronal function have not been defined in vivo Problems with the amyloid hypothesis Another concern is that the fact that all AD causing mutations in APP, PS1, or PS2 increase A-beta deposition, yet the degree to which a particular mutation affects A-beta production in cell culture shows no simple correlation with the age at which if first produces symptoms The degree of dementia appears to correlate with soluble A-beta species. Several lines of evidence have converged recently to demonstrate that soluble oligomers of A-beta, instead of monomers or insoluble amyloid fibrils, may be responsible for synaptic dysfunction in the brains of AD patients and in animal models Calcium Hypothesis Calcium modulates many neural processes, including synaptic plasticity and apoptosis Dysregulation of intracellular calcium signaling has been implicated in the pathogenesis of alzheimer’s disease Increased intracellular calcium elicits the characteristic lesions of this disorder, including the accumulation of amyloid-beta, the hyperphosphorylation of TAU and neuronal death Every gene that is known to increase susceptibility to Alzheimer’s disease also modulates some aspect of calcium signaling Calcium Hypothesis The disruption of calcium homeostasis might be one of the principal mechanisms by which A-beta manifests its neurotoxicity A-beta has been shown to destabilize neuronal calcium homeostasis, generally leading to an increase in cytosolic calcium which can then trigger neuronal apoptosis Treatment Strategies 1. One could attempt to partially inhibit proteases that generate A-beta from APP 2. One could attempt to prevent the oligomerization of A-beta or enhance clearance from the cerebral cortex 3. An anti-inflammatory strategy based on the observation that a cellular inflammatory response in the cerebral cortex is elicited by the progressive accumulation of A-beta Treatment Strategies 4. Based on modulating cholesterol homeostasis. Chronic use of cholesterol lowering drugs have been associated with a lower incidence of Alzheimer’s disease 5. Based on the observation that A-beta aggregation is, in part, dependent on the metal ions zinc and copper. This strategy reasons that chelation of these ions in vivo may prevent Abeta deposition References 1. Price, D.L., Sisodia, S.S., and Borchelt, D.R. (1998). Genetic Neurodegenerative Diseases: The Human Illness and Transgenic Models. Science 282, 1079-1093 2. Vassar, R., and Bennet, B.D. (1998) Beta-secretase cleavage of alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 5440-5464 3. LaFerla, F.M. (2002) Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nature Reviews Neuroscience 3, 862-872 4. Gregersen, N., Bross, P., and Andresen, B.S. (2001) The role of chaperone-assisted folding and quality control in inborn errors of metabolism: Protein folding disorders. Journal of Inherited Metabolic Disorders 24, 189-212