PORPHYRINS AND HEMOGLOBIN
advertisement

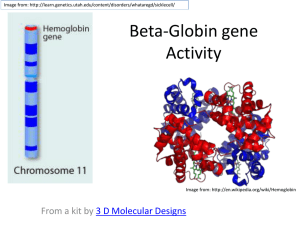
Dental Biochemistry Lectures 39 and 40 2015 HEME AND HEMOGLOBIN Michael Lea Porphyrins and Hemoglobin - Lecture Outline • • • • • • 1. Synthesis of Porphyrins 2. Regulation of Heme synthesis 3. Porphyrias 4. Degradation of Heme and Jaundice 5. Hemoglobin Structure and Function 6. Hemoglobin Pathology • Suggested reading: Lippincott’s Biochemistry 6th edition, pages 25-42, 277-285, 289 1. Porphyrin Synthesis Porphyrins serve as prosthetic groups for proteins that function in oxygen transport (hemoglobin and myoglobin), breakdown of peroxide (catalase), electron transport (cytochromes a, b and c), hydroxylation (cytochrome P450), nitric oxide synthase and light absorption (chlorophyll). The porphyrins are heterocyclic ring structures that include four pyrrole rings joined together through carbon (methenyl) bridges. The most abundant porphyrins in nature are found in hemoglobin and the chlorophylls. In the center of porphyrins a metal atom is chelated to the nitrogen atoms of the pyrrole units. In heme and related porphyrins this atom is iron. In chlorophyll the metal atom is magnesium. Structure of Heme 1. Porphyrin Synthesis The first and rate limiting reaction in porphyrin synthesis is the reaction catalyzed by 5-aminolevulinate synthase: glycine + succinyl CoA --> NH3+-CH2-CO-CH2-CH2-COO- +CO2+CoA 5 -aminolevulinate Pyridoxal phosphate is a cofactor for 5-aminolevulinate synthase (daminolevulinate synthase, ALA synthase). Synthesis of 5-aminolevulinate synthase is inhibited by heavy metal ions and there is a feedback inhibition exerted by heme. In the second reaction of porphyrin synthesis, two molecules of 5aminolevulinate condense to form the monopyrrole, porphobilinogen. The combination of four porphobilinogen molecules gives a linear tetrapyrrole and is accompanied by the loss of four molecules of ammonia . Ring closure catalyzed by uroporphyrinogen synthase and isomerization gives uroporphyrinogen III. Further intermediates in the synthesis of heme are coproporphyrinogen III, protoporphyrinogen IX and protoporphyrin IX. The conversion of protoporphyrin IX to heme is catalyzed by ferrochelatase. The first reaction and the last three reactions of heme synthesis occur in mitochondria with the other four reactions being in the cytosol. ALA synthase Porphobilinogen Synthase ALA dehydratase Ferrochelatase 2. Regulation of Heme Synthesis Regulation of hemoglobin synthesis involves control of both porphyrin and polypeptide synthesis. The regulating factor is an accumulation of heme, which in the free form is spontaneously oxidized to hemin. An accumulation of hemin diminishes the activity of 5-aminolevulinate synthase, probably by repressing synthesis of the enzyme, as well as directly inhibiting the existing enzyme, thereby shutting off porphyrin synthesis. Hemin has this effect both in the primitive red blood cell synthesizing hemoglobin and in other cells in which the cytochromes and other heme proteins are made. Hemin activates the synthesis of globin peptide by combining with an inhibitory protein. This mechanism keeps the synthesis of heme and globin in balance. 3.Porphyrias Disease Enzyme Organ Pathology Acute Intermittent Porphyria Uroporphyrinogen I synthase Neuropsychiatric deficiency Abdominal pain (porphobilinogen deaminase or hydroxymethylbilane synthase) Congenital Erythropoietic Porphyria Uroporphyrinogen III cosynthase deficiency Porphyria Cutanea Tarda Uroporphyrinogen decarboxylase Skin Hereditary Coproporphyria Coproporphyrinogen oxidase Skin, Abdominal pain, Neuropsychiatric Variegate Porphyria Protoporphyrinogen oxidase Skin, Abdominal pain, Neuropsychiatric Protoporphyria Ferrochelatase Skin, Abdominal pain, Neuropsychiatric Lead poisoning Inhibition of ALA dehydratase and ferrochelatase Nervous system, blood etc. Skin 4. Degradation of Heme The average life span of normal human erythrocytes is approximately 120 days. Senescent red cell are processed by the reticuloendothelial system. Hemin, protoporphyrin and albumin bound hemin are taken from the plasma largely by hepatic parenchymal cells and converted almost quantitatively to bilirubin. In hemolytic anemia, significant hemoglobinemia is unusual yet conjugated hyperbilirubinemia frequently occurs. This indicates that the rate of heme degradation may exceed the maximum rate of bilirubin removal by the liver. When hemoglobin is catabolized the apoprotein is degraded to the constituent amino acids and the heme prosthetic group is opened and subject to oxidation to yield biliverdin, iron and carbon monoxide. The iron is reutilized. Biliverdin is reduced to bilirubin by biliverdin reductase. Bilirubin is transferred to the liver as a bilirubin-albumin complex. The bilirubin is made more soluble by conjugation with glucuronic acid from UDP-glucuronic acid in a reaction catalyzed by glucuronate transferase. Jaundice is defined as the clinical syndrome associated with elevation of the serum bilirubin level above the normal level of 1 mg/dl. Types of Jaundice (Icterus) 1. Prehepatic jaundice (hemolytic jaundice) a. acute hemolytic anemia b. neonatal physiologic jaundice c. chronic hemolytic anemia 2. Hepatic jaundice a. conjugation failure: neonatal physiologic jaundice, CriglerNajjar disease, Gilbert's syndrome, b. bilirubin transport disturbances: Dubin-Johnson disease c. diffuse hepatocellular damage or necrosis d. intrahepatic obstruction 3. Post-hepatic jaundice Obstruction of the common bile duct by stones, neoplasms or spasms. 5. Hemoglobin Structure and Function Suggested reading: Lippincott’s Biochemistry 6th edition, pages 25-42 5. Hemoglobin Structure and Function Oxygen transport is mediated by two heme proteins, myoglobin and hemoglobin. Myoglobin occurs in muscle and has a single polypeptide chain. Hemoglobin occurs in red blood cells and has four polyeptide chains. Hemoglobin A is the major type in adults and has two alpha and two beta chains. Each subunit has a hydrophobic pocket containing the heme unit. The oxygen binding curve for myoglobin is a rectangular hyperbola whereas the oxygen binding curve for hemoglobin is sigmoidal in shape. Myoglobin has a greater affinity for oxygen than hemoglobin. The sigmoidal curve for oxygen binding to hemoglobin is an indication of the cooperativity which exists in the binding of the four oxygen molecules. The binding of oxygen to hemoglobin is decreased by a decrease in pH (Bohr effect) and by an increase in the level of 2,3-bisphosphoglycerate. 5. Hemoglobin Structure and Function Unlike the iron in cytochromes which is alternately reduced and oxidized as part of the mechanism of action, the iron in hemoglobin must be maintained in the reduced state for the transport of oxygen. Oxidation of the iron of hemoglobin from the Fe2+ to the Fe3+ state results in the formation of methemoglobin. Oxidation of hemoglobin to methemoglobin can be caused by a number of agents including nitrite and some drugs. The reverse of this process can be achieved by several NADH or NADPH-linked enzymes. Cooperativity in Oxygen binding to hemoglobin The sigmoid curve for the association of oxygen with hemoglobin reflects the cooperativity that occurs and involves two conformational states. The two conformational states are designated T (tense) and R (relaxed). Binding of oxygen to one of the four subunits favors conversion from the T to the R state where the R state has a higher affinity for oxygen. 6. Hemoglobin Pathology 1. Glucosylation of hemoglobin occurs in diabetes mellitus and may serve to measure chronic elevation of blood glucose levels. 2. One of the most important hemoglobinopathies of genetic origin is sickle cell anemia. This occurs when the individual is homozygous for hemoglobin S in which there is a point mutation resulting in a substitution of valine for glutamate in the beta subunits. There is hemolytic anemia, painful crises and poor circulation. 3. The thalassemias are a group of hereditary diseases in which there are defects in the synthesis of either the alpha or the beta chains. 4. Carbon monoxide binds to hemoglobin more tightly than oxygen and it prevents oxygen release. Hemoglobin Learning Objectives (taken from Baynes and Dominiczak) You should be able to 1. Describe the mechanism of oxygen binding to myoglobin and hemoglobin. 2. Describe the conformational differences between deoxygenated and oxygenated hemoglobin. 3. Define the concept of cooperativity in oxygen binding to hemoglobin. 4. Describe the Bohr effect and its role in modulating the binding of oxygen to hemoglobin. 5. Explain how 2,3-bisphosphoglycerate interacts with hemoglobin and influences oxygen binding. 6. Summarize the processes by which carbon dioxide is transported from peripheral tissues to the lungs. 7. Describe the major classifications of hemoglobinopathies. 8. Describe the molecular basis of sickle cell disease.