Species 3
advertisement

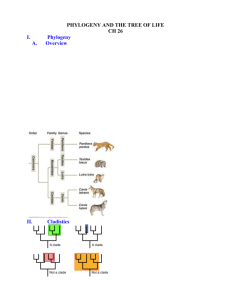
Systematics: 1) Taxonomy: Classification and naming of organisms a. Hierarchical nomenclature with taxonomic categories (kingdom, phylum, class, order, family, genus, and species) 2) Phylogenetic analysis: The study of evolutionary relationships among species a. Under common decent, hierarchical classification reflects true genealogical relationships Tree of Life http://tolweb.org/tree/phylogeny.html Phylogenetic Analysis Species 1 Species 2 Species 3 a b c d e f g h i j 0 0 1 1 1 0 0 0 0 0 a b c d e f g h i j 0 0 1 0 0 1 1 1 1 0 a b c d e f g h i j 0 0 1 0 0 1 1 0 0 1 i1 h1 e1 d1 g1 Species 4 a b c d e f g h i j 1 1 0 0 0 0 0 0 0 0 j1 Ancestor 3 (node) f1 b1 shared characters a1 Ancestor 2 (node) 1 2 3 4 c1 Ancestor 1 a b c d e f g h i j 0 0 0 0 0 0 0 0 0 0 1 1 1 0 2 4 3 0 3 5 7 0 4 5 3 4 - shared derived characters Phylogenetic Terms Species 2 Species 1 i1 e1 d1 f1 h1 g1 Species 3 Species 4 j1 Ancestor 3 Ancestor 2 b1 a1 c1 Ancestor 1 monophyletic group: set of species that share a common ancestor. synapomorphy: shared derived character state. autapomorphy: uniquely derived character state. The identification of synapomorphies help define nested series of monophyletic groups. Phylogenetic Analysis Species 1 Species 2 Species 3 a b c d e f g h i j 0 1 1 0 0 0 1 1 0 1 a b c d e f g h i j 0 1 1 1 1 1 0 1 0 0 a b c d e f g h i j 0 1 1 1 1 1 1 0 1 1 j0 h1 h1 f1 g1 d1 g1 Species 4 a b c d e f g h i j 1 0 0 0 0 0 0 0 0 0 i1 Ancestor 3 e1 shared characters a1 Ancestor 2 j1 c1 b1 Ancestor 1 1 2 3 4 1 3 4 0 2 5 5 0 3 5 6 0 4 4 3 4 - shared derived characters Phylogenetic Terms Species 1 h1 g1 Ancestor 2 Species 2 Species 3 Species 4 i1 j0 h1 g1 f1 Ancestor 3 e1 d1 j1 c1 b1 a1 Ancestor 1 homoplasy: when two species share a derived character state because of convergent evolution or evolutionary reversal, but not because of common descent. convergent evolution: independent evolution of a derived character state in two or more taxa. Types of Homoplasy Convergence: Shared derived similarities, that are not based on common origin (i.e. homology ), but on an independent origin in different taxa. Example: Wings in insects, birds, and bats Reversal :The secondary presence of an apparently ”ancestral” character state. Example: Aquatic mode of life for fish, terrestriality for tetrapods, reversal to aquatic life in whales Homoplasy: Common in DNA sequence data. Each nucleotide position defines a separate character Homoplasy - independent evolution • Loss of tails evolved independently in humans and frogs - there are two steps on the true tree Lizard Human TAIL (adult) Frog Dog absent present Homoplasy: Misleading evidence of phylogeny • If misinterpreted as a synapomorphy, the absence of tails would be evidence for a wrong tree: grouping humans with frogs and lizards with dogs Human Lizard TAIL Frog Dog absent present Homoplasy: Reversal • Reversals are evolutionary changes back to an ancestral condition • As with any homoplasy, reversals can provide misleading evidence of relationships 1 True tree 2 3 4 5 6 7 8 Wrong tree 9 10 1 2 7 8 3 4 5 6 9 10 So how do we construct trees with a sample of homologous characters? • How do we sort out phylogeny from a mixture of signal (synapomorphies) and noise (homoplasy). • Cladistic methodology (Willi Hennig) utilizes the principle of parsimony. • Parsimony= The tree that requires the fewest number of evolutionary changes or steps to explain the data is preferred. Tree Reconstruction with Parsimony Tree Reconstruction with Parsimony Tree 1 Nucleotide substitution = evolutionary step Tree 2 Tree 3 Tree Reconstruction with Parsimony Tree Reconstruction with Parsimony Character 2 I(A) Tree 1 II(G) III(A) I(A) Tree 2 IV(G) III(A) II(G) I(A) IV(G) Tree 3 IV(G) or I(A) II(G) II(G) III(A) or III(A) I(A) II(G) IV(G) IV(G) III(A) What to do when some characters tell you one thing and others tell you something else (Homoplasy)? Parsimony with Multiple Characters 1 The most parsimonious pattern of character change is noted for each character separately, for each tree. 2 The number of changes is summed across characters for each tree. 3 The preferred tree is the one that implies the fewest overall character changes. Tree 1 Tree 2 Tree 3 Tree 1 is favored under the criterion of Parsimony Parsimony • Advantages: – Simple method - easily understood operation – Does not seem to depend on an explicit model of evolution – Should give reliable results if the data are well structured and homoplasy is either rare or widely (randomly) distributed on the tree • Disadvantages: – Doesn’t always provide the best estimate of phylogeny • Maximum likelihood • Bayesian analysis Parsimony is Computationally Intensive • The number of possible trees increases exponentially with the number of species, making exhaustive searches impractical for many data sets. • Need to utilize a way to search for the best tree without evaluating all possible trees. A D E F B G E A C • Tree bisection and reconnection C A B G F D F B G D C E What if there is a large amount of homoplasy in the data? • Sequence data may have multiple, “hidden” substitutions. • Use a model of evolution to correct for different rates of substitutions or unequal base frequencies or other parameters. • Maximum-likelihood phylogenetic analysis Seq 1 Seq 2 Seq 1 Seq 2 C C T C AGCGAG GCGGAC A A Plot of base pair differences between pairs of mammalian species for a representative gene. L = P (DT, M) C A A Example: Model of sequence evolution • Simplest Model = JukesCantor - Assumes all substitutions are equally likely (a a A a a a a C G a T Example: What is the total number of substitutions? Expected Difference AGATCG CAACGC CCGGAC TTCTTA ATCGGG K = - 3 ln ( 1 4 4 p 3 ) = 0.27 total observed = 7 ; p = 7/30 = 0.23 Total expected = 0.27 x 30 = 8.24 Sequence Difference AGGTCG CATTGC CCCGAT CTCTTG ATCGGG Correction Observed Difference Time Phylogenetic Inference Using Maximum Likelihood • Model of sequence evolution and the estimation of its parameters allows the placement of probabilities on different types of substitutional change. • Likelihood analysis focuses on the data, not the tree. It is the Probability of the Data given a Tree and a Model of evolution. Seq 1 Seq 2 ATATC CTAGC L = P (DT, M) The Likelihood (i.e. the probability of observing the data) is a sum over all possible assignments of nucleotides to the internal nodes Phylogenetic Inference Using Maximum Likelihood • Calculate the Likelihood for each base position in the sequence and summarizes across all base positions. • The ML tree is the tree that produces the highest likelihood. • Evaluates the branching structure of the tree, and also the branch length, using similar tree-searching strategies as used in parsimony analysis. – This is important, because by using a model-based approach, mutational change is more probable along longer branches than on shorter branches. • Can be extremely computationally intensive. Phylogenetic Inference Using Maximum Likelihood • Important point about ML: The model you choose to use can have a large impact on the resulting ML tree. • If you flip a coin and get a head, what is its likelihood? – If it’s a 2 sided and fair coin (your model), the likelihood is 0.5 – If it’s a two-headed coin (your model), the likelihood is 1.0 Assessing the Robustness Of Trees • We can use a number of methods to assess the robustness of particular branches in our trees – Bootstrapping (Jackknifing, Decay-Index) •Bootstrapping: • Multiple new data sets are made by re-sampling from the original data set. –Bootstrapping: Sampling done with replacement • The resampled data sets are subjected to phylogenetic analysis. • The proportion of times a clade appears in the trees across all replicate data sets is called its bootstrap proportion. QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. Taken from Baldauf, S. L. Phylogeny for the faint of heart: a tutorial. Trends in Genetics 19:345-351. Bootstrapping • Clades that receive a high bootstrap are considered to be more supported by the data than clades with a lower bootstrap. – 70% or greater is good, but many phylogeneticists will only consider branches with ≥90% as being strongly supported. Bootstrap • Can perform with any type of phylogenetic analysis: parsimony, ML, distance-based • Important to emphasize that a bootstrap does not reveal the probability that a particular clade is true, but only how well it is supported by the particular dataset. Molecular Clocks • The mutation rate for some genes may be relatively constant across species. • This idea is based on neutral theory (this will be introduced later in the course) - nucleotide or amino acid substitutions occur at a rate equal to the mutation rate. • Generally in applying a molecular clock, you assume that the mutation rate for a gene does not differ among species. Molecular Clocks 1) Construct A Tree 2) Date a Node in the Tree Outgroup Outgroup Species 1 Species 1 Species 2 Species 2 Species 3 Species 3 Species 4 Species 4 Fossil for Species 4 ~1 MY 3) Calculate Divergence Species 3 Species 4 2% Sequence Divergence } You know that the most recent possible divergence between 3 and 4 is at least 1 MY 4) Calculate a Rate R= 2%/1MY Molecular Clocks 5) Apply Rate to Other Nodes in Tree Outgroup Species 1 Species 2 Species 3 5MY Species 4 2MY 1MY • Best applied when dates available for multiple nodes. • Can utilize solid geological information as well as fossil information. • Must be aware of possible non-clock behavior of genes. Phylogeny of North American Black Basses Near et al., 2003. Evolution 57:1610–1621. Previous hypothesis that speciation within the genus Micropterus occurred during the Pleistocene. Micropterus has a very good fossil record. Calibration of a molecular clock and calculation of divergence times among species reveals that most species diverged well before the Pleistocene QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. Species Delimitation in Rapidly Radiating Systems • Accumulation of species diversity over short periods of time. • Adaptive radiations • Often of very recent origin • Difficult to resolve monophyletic species-level lineages. Salzburger, W. and A. Meyer. 2004. Naturwissenschaften 91:277-290. Species Delimitation in Rapidly Radiating Systems (Species trees vs gene trees) Lineage sorting and the retention of ancestral alleles or allelic lineages Species Delimitation in Rapidly Radiating Systems Lineage sorting and the retention of ancestral alleles or allelic lineages Darwin’s Finches East African Cichlid Fish Moran and Kornfield. 1993. Mol. Biol. Evol. 10:1015-1029. Takahashi et al. 2001. Mol. Biol. Evol. 18:2057-2066. Sato et al. 1999. PNAS. 96:5101-5106. Species Delimitation in Rapidly Radiating Systems Limited reproductive isolation leads to hybridization and introgression Ambystoma tigrinum species complex A. californiense Shaffer & McKnight 1996 Evolution 50:417-433 Gerald and Buff Corsi © California Academy of Sciences An early study found that A. ordinarium was not a monophyletic group (when using mtDNA as the source of characters). Indeed, more data shows extensive mtDNA nonmonophyly with respect to A. ordinarium. Polyphyly: A. ordinarium does not form a monophyletic group. Paraphyly: A. ordinarium does form a monophyletic group but other species should also be included in this group, based on the character that is used to reconstruct relationships. Nuclear Genes Summary • 4 genes yield A. ordinarium monophyly. • 3 genes yield A. ordinarium paraphyly. (2 are nearly monophyletic.) • 1 gene yields A. ordinarium polyphyly. • Nuclear data strongly suggests that A. ordinarium is a monophyletic lineage. Signatures of Rapid Lineage Diversification Poe, S., and A. L. Chubb. 2004. Syst. Biol. 58:404-415. Short Internal Branches Phylogenetic Discordance Among Loci A. dumerilii Shared and minimally divergent mtDNA haplotypes strongly indicate recent hybrid introgression.