Details of all metric derivations and model specifications
advertisement

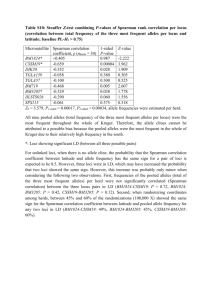
Buckee et al.: Supplementary material The D* measure of association Hedrick and Thomson [15] developed several methods for analysing linkage between two loci with multiple alleles. These are more appropriate for the Czech data than the many sophisticated methods for analysing multiple loci, because they allow for comparison between different types of genes in the same sample. Although several statistics were applied to the data, they all showed similar results and are based on similar metrics, so only one was presented. The disequilibrium between two loci is given by: Dij = xij - piqj Where xij is the observed frequency of AiBj, pi and qj are the frequencies of alleles i and j at loci A and B, respectively, such that when there is no association between loci, the expected frequency of AiBj is piqj. The total disequilibrium between all alleles at two loci may then be expressed as: k l D Dij2 2 i1 j1 These measures of association between loci are dependent on allelic frequencies, comparing exhibit different levels of diversity, a more however. Since the loci we are appropriate metric for measuring linkage disequilibrium is one that is standardized by single-locus heterozygosity. Here, we define the homozygosity (F) of loci A and B, having k and l alleles respectively, as: k FA pi2 i1 l FB q 2j j1 Adjusting these measures to consider the heterozygosities at each locus, we then use the association: following measure of two-locus D* D2 (1 FA )(1 FB ) a stronger level of association or linkage. By using D* to Higher values of D* indicate analyze data which spans over twenty-five years, the transient linkage caused by shortterm clonal spread should be eliminated, and strong associations caused by other processes will be highlighted. Note that this measure of linkage disequilibrium may not be completely independent of allele frequencies (Hedrick, 1987) but was used as a relative measure of association rather than an absolute one. We applied another statistic suggested by Hedrick (1987) with a different approach to adjusting for different allelic frequencies, to verify our findings: k l D piq j Dij i1 j1 where D’ij is defined as: Dij Dij Dmax Here, if Dij < 0 then Dmax is the smaller of piqj and (1 - pi)(1 – qj), and if Dij > 0 then Dmax is the smaller of (1 - pi)qj and (1 - qj)pi. The associations between loci using this measure were equivalent to those using D*, with the same comparisons being significantly from one another in both cases. Derivation of f* metric Under the theoretical framework of Gupta et al. (1996), antigenic loci should display non-overlapping patterns of allelic association as a result of strong immune selection. A very simple new statistic for measuring these kinds of associations was developed and tested using simulations, before applying it to the data. A non-overlapping structure should result in a matrix of allelic associations between two loci that shows a diagonal structure when ordered correctly. In other words, an allele at locus 1 should be predominantly associated with only one allele at locus 2, such that the prevalent strains should dominate both their ‘row’ and column’ of the association matrix (see Figure 1 below). Figure 1: A schematic showing a hypothetical non-overlapping distribution, where shaded squares represent the most dominant strains for different alleles. The extent of non-overlapping structure within an allelic association matrix can therefore be measured by assessing the dominance of allele combinations. For isolates characterized at two loci, A and B, let pi be the frequency of allele i at locus A, and qj be the frequency of allele j at locus B. As above, xij is then the frequency of allele combination i at locus A and j at locus B. The dominance of the most prevalent allelic combination containing the allele i at locus A and the most prevalent allelic combination containing allele j at locus B, respectively, is then given as: x ik3 f A (i) for the k that maximizes xik pi qk f B ( j) x kj3 for the k that maximizes xkj pk q j which may be seen as the “row” and “column” dominance for the matrix of allelic combinations, multiplied by the overall frequency. The sum over all alleles at locus i will therefore be close to 1 when the allelic matrix is highly non-overlapping with respect to this locus. The overall overlap between two loci is then given by the average for each of the two loci: 1 f * f (i) f ( j) A B 2 i j Thus, f* will be close to one if the allelic combinations are highly non-overlapping, and show this structure. Note that this measure could be generalized lower when they do not to include more loci. Simulations If the associations between loci were generated by means of a purely neutral, relatively random process, then one would expect an even distribution of allelic associations. Bacteria reproduce clonally, however, and the emergence and spread of successful clones means that certain combinations of alleles will dominate at any point in time. A purely random distribution of alleles would not adequately depict the distribution of alleles under neutral selection in a bacterial population, therefore. A stochastic model of strain evolution was used to generate hypothetical distributions of allelic associations and test the f* statistic. Hosts were connected in a ring lattice as described in Buckee et al. (2004) and developed further in Buckee et al. (2008), and details of the model framework may also be found in these papers. In short, the contact network of hosts was generated randomly, equivalently to the network described in Watts and Strogatz (1998) when . Here, is the probability that an individual will come into contact with a randomly chosen individual rather than a local neighbor in the ring lattice. To approximate a mean-field mixing process, the host contact network changed at every time step. Following infection by a particular strain they generated allele-specific immune memory, which protected against the antigens. Strains were defined by three loci, two antigenic and one housekeeping, each with five alleles (thus, strain space can be envisaged as a 5x5x5 matrix of allelic associations). The two antigenic loci determined host immunity, with hosts gaining a degree of protection against strains sharing alleles with a previously ‘seen’ strain dependent on γ, the level of cross-immunity. We assumed that a host’s vulnerability to infection by a strain was dependent on the similarity between that strain and the strains in the host’s immune memory, an assumption also made in Gupta et al. (1998). Given this assumption, the fraction of identical alleles between the host’s immune memory and the infecting strain was converted into a vulnerability of infection (v), between 0 and 1, using: v (1 f 1 / ) where f is the fraction of identical alleles and is a positive number scaling the level of cross-immunity. The housekeeping gene had no effect on the transmission of the strain, however it was subject to the same rates of mutation and recombination as the antigenic genes. All strains had exactly the same R0 value. Strong immune selection against overlapping antigenic associations should create the diagonal structure illustrated in the matrix above, however the associations between the antigenic loci and the housekeeping locus, as well as the associations between antigens when cross-immunity is low, should reflect the random pattern of clonal reproduction, strain transmission and recombination. Table 1 shows the range of parameters explored in the simulations, by means of Latin Hypercube Sampling (Blower & Dowlatabadi, 1994). 1500 simulations were run with different levels of cross-immunity until they had reached equilibrium, and the frequency of strains in the 5x5x5 matrix were then analysed. Note that the effects of cross-immunity were relatively unaffected by changes to other parameters explored. Figure 2 shows the frequency distribution of f* scores for simulations for antigen:antigen associations under strong cross-immunity (>2) and for antigen:housekeeping associations and antigen:antigen associations when cross-immunity is weak (<=2). Figure 2: Frequency distributions of f* scores for different types of two-locus association. The yellow bars correspond to antigen:antigen comparisons for >2, the dark blue bars correspond to both antigen:housekeeping gene comparisons, and antigen:antigen comparisons when <=2 (note that this latter group account for all simulations resulting in an f* value of 1). For the antigen versus housekeeping gene comparison, as well as for antigen versus antigen comparisons when cross-immunity was low, f* scores were low, since there was no immune selection on the housekeeping gene, and therefore no dependence on the level of cross-immunity. For antigenic loci however, as cross-immunity increased, there was increasing selection against strains that shared alleles, and a non-overlapping structure emerged. Table 1: parameter ranges explored in the simulations. LHS Range Parameter Description [Min:Max] C Mean number of contacts per host [8:20] Average duration of infection [3: 10] Average duration of immunity [10:30] Probability of transmission (to a completely susceptible host) [0.2:0.8] R Probability of recombination per allele [0.01:0.1] Probability of allelic mutation per allele [0.001:0.005] Degree of cross-immunity [0.02:4] P Host population size [500:1500] Network metrics The density of a network is simply given as the number of edges in a network, divided by the total possible edges. We use transitivity as a measure of clustering in the network. Transitivity is a triadic structural constraint measuring the proportion of triads (groups of three nodes, say a, b, and c) in the network that follow the rule: a -> b -> c => a -> c. Within the social network literature, this relationship between triplets represents the idea that “friends of friends are also friends”. A high density of transitive triads leads to a highly clustered network. Betweenness centrality is a node-level metric that is calculated for each node as the number of shortest paths between all pairs of nodes in the network that pass through it. In other words, nodes with a high betweenness are those that act as bridges between different parts of the network. The degree distribution of a network is simply a frequency distribution of the number of edges each node has.