SUPPLEMENTARY INFORMATION Reagents. SLC of TGF
advertisement

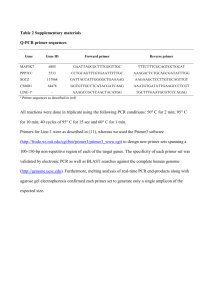
SUPPLEMENTARY INFORMATION Reagents. SLC of TGF-1, mature TGF-1 and TGF-2, TGF-1 and -2 neutralizing Abs, and isotype control Abs were obtained from R&D systems. The ELISA to detect human LAP-1 was performed with MAb clone 9005 as a capture Ab and biotinylated MAb clone 27240 for detection. The TGF- neutralizing mAb 1D11 was employed for in vitro assays and the TGF- neutralizing Ab purified from the 2G75A9 hybridoma (a gift from Dr. J. Leterio, National Cancer Institute, National Institutes of Health) was employed for the in vivo experiments. These Abs neutralize all three TGF- isoforms. Isotype controls were obtained from BioXcell. Insulin-, transferrin-, sodium selenite (ITS), and siliconized tubes were from Sigma-Aldrich. Galardin was obtained from Enzo Life Sciences and SB 431542 from Cayman Chemicals. All other protease inhibitors were obtained from Roche. Interferonγ was obtained from R&D systems and Staphylococcus aureus Cowans (Pansorbin®) from Calbiochem. Functional grade anti-CD3ε (clone 145-2C11) was obtained from eBioscience and antiCD28 (clone 37.51) from BD Pharmingen. Conditioned media (CM) was collected from confluent cells kept in serum-free conditions for 16h prior to media collection and was cleared by centrifugation prior to quantitation of TGF- by ELISA specific for the isoform indicated. RNA isolation and quantitative PCR analysis. Total RNA was extracted from colon tissue using the Nucleospin RNA/protein isolation kit (Macherey-Nagel). After DNase digestion, cDNA was generated using random primers and Superscript II under the manufacturer’s recommendations (Invitrogen). Real-time qPCR was performed on the TaqMan 7500 System (Aplied Biosystems). For each reaction, 1 µL cDNA, synthesized from 1 µg RNA in 25 µL, was used in a total volume of 11 µL containing 6.25 µL of Power SYBR Green PCR master mix (Applied Biosystems), 3.75 µL water and 450 nM of the appropriate forward and reverse primers. The following primers were used: IFN-γ (forward primer: 5’-TAGCCAAGACTGTGATTGCGG-3’, reverse primer: 5´-AGACATCTCCTCCCATCAGCAG-3’); TNF-α (forward primer: 5’-AAGCCTGTAGCCCACGTCGTA-3’, reverse primer: 5´-AGGTACAACCCATCGGCTGG3’); IL-17 (forward primer: 5’-TCCAGAAGGCCCTCAGACTA-3’, reverse primer: 5´- AGCATCTTCTCGACCCTGAA-3’); TGF-β1 (forward primer: 5’-TTGCTTCAGCTCCACAGAGA-3’, reverse primer: 5’-TGGTTGTAGAGGGCAAGGAC-3’) and IL-10 (forward primer:5´-CAGAGCCACATGCTCCTAGA3´, reverse primer: 5´-TGTCAGCTGGTCCTTTGTT-3´). The PCR profile was as follows: 2 min 50°C, 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 60 sec at 60°C. Subsequently, a melting curve analysis was performed which consisted of 70 cycles of 10 sec with a temperature increment of 0.5°C/cycle starting at 60°C. The relative expression was calculated according to the equation Rel. Exp (RE)= 2-ΔCt. Protein production and purification. PSG1-Fc, PSG1-His-FLAG, and the control proteins were harvested from the supernatant of transiently or stably transfected cells and purified as previously reported (30). PSG1 was purified from sera pooled from pregnant women and is referred as native PSG1. To obtain native PSG1, pregnant serum was pre-cleared on a protein G column (Thermo Scientific) after which, it was passed through an anti-PSG1 mAb #4-Sepharose affinity column. PSG1 was eluted with 0.1M glycine [pH 2.9] and immediately neutralized with 100μl of 1M Tris-HCl [pH 8.0]. Individual fractions were analyzed by Western blot with the anti-PSG mAb BAP3 (GenoVac) and positive fractions were pooled, concentrated, and buffer exchanged with PBS using Amicon Ultra-15 10-KDa MWCO centrifugal filter units (Millipore). To quantitate and assess the purity, native PSG1 was separated on a 4-12% NuPAGE Bis-Tris gel (Invitrogen) and stained with GelCode Blue (Thermo Scientific). Native PSG1 was quantitated with anti-PSG1 mAb#4 against recombinant PSG1-Fc, used as standard, by immunoblot analysis of different dilutions of both proteins. Luciferase and SEAP Assays. Proteins were added to the reporter cells in a final volume of 100 μl in serum-free DMEM with 0.1% ITS (Sigma-Aldrich) for the PAI-1 luciferase reporter mink lung epithelial cell (TMLEC) or in serum-free DMEM for the MFB-F11 cells. The TGF-β reporter HEK-SMAD cells were generated by transduction of the Cignal Lenti SMAD reporter lentivirus as recommended by the manufacturer (SABiosciences). Cells were selected with 1 μg/ml puromycin after which single cell clones were generated by limited dilution. Single clones (1.6 x 104 cells per well of a 96 well polyL-lysine coated plate) were seeded in 100μl of DMEM-10% FBS for 2h, after which the media was removed and replaced with DMEM-0.1% ITS containing different concentrations of mature recombinant TGF-β1. Clones 8 and 10 were determined to be the most sensitive in responding to TGF-β1 and were selected for further studies showing similar results upon treatment with PSG1. For the three reporter cell lines, we constructed a calibration curve using known amounts of mature TGF-β1 or -β2 to quantify their response to the active cytokine. The luciferase and SEAP activity values obtained upon incubation of the reporter cells with serum-free DMEM with 0.1% ITS or DMEM only (for MFB-F11 cells) was used as blank, and was subtracted from the reported values. FACS analysis. For the detection of cell membrane bound LAP, cells were dislodged from flasks with Accutase (Innovative Cell Technologies) and 105 cells in 100 μl of FACS buffer (2% BSA, 0.02% NaN3 in PBS) were incubated on ice with PE conjugated-anti-human LAP (clone 35409) or a PE-conjugated isotype control (Serotec). Surface expression of PSG1 in HeLa-PSG1 cells was confirmed with the anti-PSG1 BAP3 mAb followed by PE-conjugated anti-mouse IgG. Cells were analyzed in the BD LSRII and fifty thousand events were collected using the BD FACS DIVA software. Post-acquisition analysis was performed with the FlowJo software (Tree Star, Inc). Isolated LPMC were resuspended in PBS, 1% Bovine serum albumin (BSA) solution and pre-incubation with 1% anti-mouse CD16/CD32 monoclonal antibody and 10% mouse serum was carried out to avoid unspecific staining via Fcreceptor. Then LPMC were incubated for 30 min at 4°C with PerCP conjugated rat anti mouse CD4 (clone RM4-5, BD Biosciences) and FITC- anti-human LAP/TGF-β1 (BAF-246, R&D systems). Subsequently, LPMC were washed and fixed/permeabilized, using FACS Fix/Perm Solution (eBioscience). PE conjugated rat anti-mouse Foxp3 antibody (clone FJK-16s, eBioscience) was incubated for 30 min at 4°C. After washing the acquisition was performed using the FACSCalibur system (BD Biosciences). Data were analyzed using FlowJo software. Cytokine determination by cytometric bead arrays (CBA). Cytokines were analysed in cell culture supernatants using cytometric bead arrays (CBA). Briefly, 48h culture supernatants were harvested and stored at -80°C until cytokine determination. IL-12p40, TNF-α, IFN-γ, IL-6 and IL-10 were detected simultaneously using the mouse inflammation cytokine CBA kit (BD Biosciences, San Diego, CA). Briefly, 50 µl of each sample was mixed with 50 µl of mixed capture beads and 50 µl of the mouse inflammation PE detection reagent consisting of PE-conjugated anti-mouse IL-12p40, TNF-α, IFN-γ, IL-6 and IL-10. The samples were incubated at room temperature for 2h in the dark. After incubation with the PE detection reagent, the samples were washed once and resuspended in 300 µl of wash buffer before acquisition on the FACSCalibur (Becton Dickinson). Data were analyzed using FCAP array software (BD Bioscience). ELISAs. To study the PSG1-mediated activation of TGF-β1 in a cell-free system, the small latent complex (SLC) at 0.5nM or 1nM as indicated in the figure legend, and different concentrations of PSG1 or control proteins were incubated together or alone for 1h at 37oC in a final volume of 100μl of PBS in siliconized tubes. The samples were transferred to blocked wells of Nunc Maxisorb plates that had been coated with capture antibody that recognizes all three TGF-β isoforms or with 1.5μg/ml of recombinant human TGF-β RII-Fc (R&D systems). After 2h, the plates were washed and the presence of active TGF-β1 was detected with biotinylated detection anti-TGF-β1 Ab, which is specific for mature TGF-β1, from the DuoSet ELISA kit (DY240). TGF-β DuoSet ELISA kits specific for each TGF-β isoform were obtained from R&D systems and the assays performed under the manufacturer’s recommendations. For activation of latent TGF-β, samples were acid activated followed by neutralization as recommended in the TGF-β DuoSet ELISA kit. Isolation of lamina propria (LP) and mesenteric lymph nodes (MLN) cells. LP cells were prepared from freshly isolated large intestine. To this end, intestinal segments were incubated twice with Hank’s balanced salt solution (HBSS) containing DTT (0.15 mg/ml) and EDTA (0.1mM) for 15 min at 37°C with agitation. After removal of the epithelial layer by decantation, the intestinal segments were incubated with RPMI 1640 containing 2.5% fetal calf serum (FCS), Collagenase (300 mg/ml, Sigma Aldrich) and DNase I (50 mg/ml, Sigma Aldrich) three times for 30 min each at 37°C with gently agitation in a CO2 incubator. Cell pellets were collected in a tube through a 40μm net (BD Biosciences) with RPMI+5%FCS and mononuclear cells were purified by Lympholyte-M (Cedarlane Laboratories) gradient centrifugation. These cells were used to characterize surface expression by flow cytometry. Lymphocytes from mesenteric nodes were isolated by smashing tissues and red blood cells were lysed with Ammonium chloride (ACK) buffer (155 mM of NH4Cl, 10 mM KHCO3 and 1 µM of EDTA). MLN cells were cultured at a concentration of 106 cells/ml with or without stimulation by plate-bound anti-CD3ε (10μg/ml) and soluble anti-CD28 (1μg/ml) for 48h. Alternatively, MLN cells were stimulated overnight with IFN-γ (1000 U/ml) and then with Staphylococcus aureus Cowans (SAC) at a 1:10000 dilution for an additional 24h. Table S1 TGF-β1 and TGF-2 associated with recombinant and native PSG1 Cell line /Origin CHO-K1 Protein produced PSG1-Fc Concentration tested TGF-β1 (active/total TGF-β2 (active/total (μg/ml) pg/ml) pg/ml) 30 N.D./ 315.3 ±11.3 N.D./N.D. 60 71.1±2.8/ 673.0±9.7 N.D./N.D. CHO-K1 PSG1-His30 206.4±7.9./ 479.7 ±3.3 N.D./N.D. FLAG 60 489.1±7.6/ 726.2±18.0 N.D./N.D. HEK-293T PSG1-Fc 30 85.1±6.9/ 335.3±6.1 N.D./N.D. 60 250.1±6.4/ 684.3±9.8 N.D./N.D. HeLa PSG1-Fc 30 N.D./ 143.0±0.5 N.D./N.D. 60 34.2±3.1/ 366.7±7.1 N.D./N.D. MEF TGF-β1- PSG1-Fc 30 N.D./N.D. N.D./98.2±4.8 null 60 N.D./N.D. N.D./ 146.1±2.5 Pregnant nPSG1 30 130.9±1.4/ 370.2±3.3 N.D./104.7±1.0 sera 60 301.3±5.1/ 776.1±4.1 N.D./ 199.4±13.5 Abbreviations: Pregnancy-specific β-glycoprotein 1 (PSG1) native PSG-1 (nPSG1), non-detectable (N.D.) Table S2. Absolute numbers of cells isolated from the mesenteric lymph nodes (MLN) of DSS-treated mice Group Abs. Nr. (x106) Flag-Fc + Isotype control 5.98 ± 0.89 Flag-Fc + α-TGF-β 5.12 ± 0.62 PSG1 + Isotype control 4.20 ± 1.02 PSG1 + α-TGF-β 4.68 ± 0.78 Data are presented as mean ± SEM derived from five mice per group.