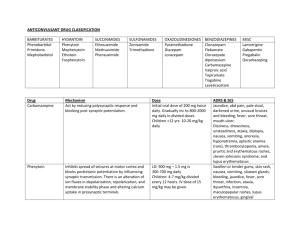
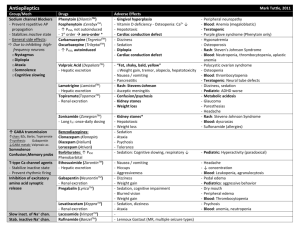
3rd. An example to illustrate drug interaction of valproic acid
advertisement

Ajman University of Science & Technology Faculty of Pharmacy & Health Sciences Clinical Pharmacokinetics Of Valproic Acid Prepared by: Dana Krayem 9860367 Supervised by: Dr. Rafiq Abou Shaaban Back Contents Chemical structure Commonly used brand names Mechanism of action Pharmacokinetics (ADME) Drug interactions: A. Drugs that affect valproic acid B. Effect of valproic acid on other drugs 3rd. An example to illustrate drug interaction of valproic acid Time to sample Adverse effects Supplied Questions & Answers Valproic acid Chemical Structure: CH3 CH2 CH2 CH CH3 CH2 COOH CH2 Commonly used brand names are: Depakene Depacon Depakote Depakote Sprinkle Mechanism of Action: Valproic acid is a branched-chain fatty acid. Its chemical structure differs significantly from any other anticonvulsant in current use. As with other anticonvulsants, the mechanism of action of valproic acid is not fully understood. It has been theorized that valproic acid acts by increasing the concentrations of the inhibitory neurotransmitter gammaaminobutyric acid (GABA) within the central nervous system through inhibition of GABA degradation or enhancement of GABA synthesis and release. Other research has suggested that valproic acid acts via inhibition of excitatory neurotransmitters or by action at sodium and calcium channels to reduce sustained (high frequency repetitive) neuronal firing. Pharmacokinetics: Absorption The time to reach maximum serum concentrations differs among the products available. Peak concentrations occur approximately one to three hours following administration of the capsule or liquid dosage forms. Peak concentrations are delayed for up to three to five hours with the sprinkle and delayed release tablet formulations. Administration of valproic acid products with food delays the rate of absorption, but does not typically affect the extent of drug absorbed. Also absorption is delayed when drug is administered in the form of enteric-coated tablets. Distribution Valproic acid is rapidly distributed throughout the body and is highly protein bound (~90%). The binding depends on the concentration of binding proteins (i.e. albumin) and binding modulators (e.g. free fatty acids). As a result, significant protein loss may cause more free drug to be available, increasing clinical effect and the potential for toxicity. Although not routinely used in patient monitoring, free valproic acid serum concentrations can be measured to assess changes in protein binding. Metabolism Valproic acid undergoes extensive hepatic metabolism. The primary metabolic pathways include both glucuronidation and oxidation, resulting in the formation of at least five different metabolic products. The 2-en-valproic acid metabolite may be pharmacologically active, while the 4-en-valproic acid may be involved in the hepatotoxicity associated with this drug. Elimination Only 1 to 3% of a dose is excreted in the urine unchanged. The usual elimination half-life of valproic acid in adults and children greater than 10 years of age is 9 to 16 hours. Younger children have a slightly shorter half-life of 7 to 13 hours, while infants have a prolonged elimination (17 to 40 hours). The clearance of valproic acid positively correlates with the unbound concentrations and is strongly age-dependent, being low in neonates and high at the end of the first postnatal month, and progressively decreasing from 2 months to 14 years. Key Parameters Therapeutic plasma concentration Bioavailability (F) Salt form (S) Volume of distribution (Vd) 50-100 mg/L 100 % 1 0.14 (0.1 – 0.5 ) L/kg Clearance (Cl) Children Adults 13 mL/kg/hr 8 mL/kg/hr Half-life (t 1/2) Children Adults Children < 10 days Cirrhoses or acute hepatitis 6 – 8 hr 10 – 12 hr 10 – 67 hr up to 18 hr Serum peak levels Capsules & syrups With food Enteric–coated tablets (Divalproex Na) 1 – 4 hr 6 – 8 hr 3 – 4 hr Drug interactions: A – Drugs that affect valproic acid (VPA): Drug Effect Phenobarbital Inc. VPA clearance Phenytoin Inc. VPA clearance Carbamazepine Inc. VPA clearance Primidone Inc. VPA clearance Rifampin Inc. VPA oral clearance Thorazine Dec. VPA clearance Cimetidine Dec. VPA clearance Zidovudine Inc. AUC Chlorpromazine Inhibits VPA metabolism Fluoxetine Inhibits VPA metabolism MAOIs Inhibits VPA metabolism Salicylates (eg.Aspirin) Displaces VPA PPB Charcoal Dec. VPA absorption B – Effect of valproic acid (VPA) on other drugs: Drug Effect Phenobarbital Inhibits metabolism of phenobarbital Carbamazepine Inhibits metabolism of carbamazepine Ethosuximide Inhibits metabolism of ethosuximide Felbamate Inhibits metabolism of felbamate Lamotrigine Reduces metabolism of lamotrigine Phenytoin Displaces phenytoin PPB Warfarin Displaces warfarin PPB Clozapine Displaces clozapine PPB Midazolam Displaces midazolam PPB Diazepam Displaces diazepam PPB Lorazepam Reduces lorazepam plasma clearance An example to illustrate drug interactions of valproic acid is: Effect of valproate on the pharmacokinetics and pharmacodynamics of lorazepam: The pharmacokinetic-pharmacodynamic interaction between valproate and lorazepam was evaluated in this randomized, double-blind, placebo-controlled crossover study. Sixteen healthy male volunteers enrolled in the study to receive either divalproex sodium (500 mg every 12 hours) or matching placebo for 12 days in the first period, and then to receive the other regimen for an identical second 12-day period. In both periods, lorazepam (1 mg every 12 hours) was administered on days 6 through 9 and on the morning of day 10. Concomitant administration of divalproex sodium with lorazepam resulted in an 8%, 20%, and 31% increase in steady-state maximum plasma concentration, area under the concentration-time curve, and trough plasma concentrations of lorazepam, respectively. The apparent clearance of lorazepam through the formation of lorazepam glucuronide was reduced by 31% during coadministration of divalproex sodium. Pharmacokinetic properties of valproate did not change significantly in the ten available participants during coadministration of lorazepam. Sedation scales revealed no statistically significant differences in sedation between the two regimens. It is concluded that valproate increases plasma concentrations and reduces clearance of lorazepam, most likely by impairing hepatic glucuronidation, and that coadministration of lorazepam does not affect the steady-state pharmacokinetic properties of valproate. Time to Sample : Due to wide fluctuations in plasma valproate concentration within a dosing interval, monitoring both the peak and trough concentrations of this drug would seem to be desirable. Nevertheless, only trough concentrations are routinely monitored because of the uncertainly about the time at which peak plasma concentrations will occur and the difficulty evaluating markedly elevated valproic acid concentrations given its capacity-limited plasma protein binding. In general, valproic acid concentrations in plasma are monitored within two to four days following: 1) initiation of therapy; 2) change in a dosing regimen; or 3) addition of other antiepileptic drugs to the patient’s regimen. Valproic acid concentrations are also measured whenever the patient’s clinical course has changed (e.g.,a decrease in seizure control or laboratory or physical findings consistent with valproic acid toxicity). As stated previously, there is some evidence that even though valproic acid concentrations may have plateaued relatively rapidly, increased therapeutic effects may continue to be observed for some time following the achievement of steady-state valproic acid concentrations. Adverse Effects Gastrointestinal: Nausea, vomiting and indigestion are the most commonly reported side effects at the initiation of therapy. These effects are usually transient and rarely require discontinuation of therapy. Diarrhea, abdominal cramps and constipation have also been reported. Anorexia with some weight loss and increased appetite with some weight gain have also been observed. CNS Effects: Sedative effects, ataxia, headache, nystagmus, diplopia, asterixis, "spots before the eyes", tremor, dysarthria, dizziness and incoordination. Rare cases of coma have been reported in patients receiving valproic acid alone or in conjunction with phenobarbital. Dermatologic: Transient increases in hair loss , skin rash and petechiae have been noted. Endocrine: Irregular menses and secondary amenorrhea in patients receiving valproic acid. Abnormal thyroid function tests have been reported Psychiatric: Emotional upset, depression, psychosis, aggression, hyperactivity and behavioural deterioration . Musculoskeletal: Weakness Hematopoietic: Thrombocytopenia has been reported. Valproic acid inhibits the second phase of platelet aggregation. This may be reflected in altered bleeding time. Bruising, hematoma formation and frank hemorrhage have been reported. Relative lymphocytosis and hypofibrinogenemia have been noted. Leukopenia and eosinophilia have also been reported. Anemia and bone marrow suppression have been reported. Hepatic: Minor elevations of transaminases and LDH, increases in serum bilirubin and abnormal changes in other liver function tests. These results may reflect potentially serious hepatotoxicity Metabolic: Hyperammonemia & hyperglycinemia Pancreatic: Acute pancreatitis Others: Edema of the extremities Supplied Capsules: 250mg: Each orange-colored, soft gelatin capsule contains: Valproic acid 250 mg. Also contains methyl- and propylparaben. Alcohol-free, gluten-free, lactose-free, sucrosefree, sulfite-free and tartrazine-free. Bottles of 100 and 500. 500mg: Each pale yellow, oval, soft gelatin enteric coated capsule contains: Valproic acid 500 mg. Also contains methyl- and propylparaben and tartrazine. Alcohol-free, glutenfree, lactose-free, sucrose-free and sulfite-free. Bottles of 100 and 500. Syrup: Each 5 mL of red syrup contains: The equivalent of 250 mg valproic acid, as the sodium salt. Also contains methyl- and propylparaben and sucrose. Energy: 74.08 kJ (17.70 kcal)/5 mL. Alcohol-free, gluten-free, lactose-free, sulfite-free and tartrazinefree. Bottles of 450 mL. Questions & Answers Question # 1 An eight-year-old, 25kg male, is receiving 250 mg of valproic acid Q 12 hr for absence seizures. While the seizure frequency has declined with this therapy, he is still experiencing one to two absence seizures per day. He has experienced no obvious side effects from the valproic acid therapy and has normal renal and hepatic function. What is the expected trough concentration for him on his current regimen? Answer: Cl = (13 mL/kg/hr) (25kg) =325 mL/hr or 0.325 L/hr Vd = (0.14 L) (25 kg) = 3.5 L Cl Kd = Vd 0.325 L/hr = 3.5 L = 0.093 hr-1 (S) (F) (Dose) Cpssmin = (e-Kdt) Vd (1 – e-Kdt) (1) (1) (250) (e-(0.093) (12) ) = (3.5) (1 – e-(0.093) (12)) = 34.8 mg/L ~ 35 mg/L Question # 2 He had a measured trough concentration of 25 mg/L. Because of the inadequate seizure control and the lack of apparent side effects, it is decided to increase the trough concentration to 50 mg/L. What dose will be required to achieve the target trough concentration of 50 mg/L if the dosing interval is decreased from Q 12 hr to Q 8 hr? Answer: (S) (F) (Dose) Cpssmax = [ Cpss min ] + Vd (1) (1) (250) = [25 mg/L] + 3.5 L = 96.4 mg/L ln (Cp1/Cp2) Kd = t ln (96/25) = 12 = 0.112 hr-1 (S) (F) (Dose) Cpssmin (e-Kdt) = Vd (1 – e-Kdt) ( Cpssmin ) (Vd) (1 – e-Kdt) Dose = (S) (F) (e-Kdt) (50) (3.5) (1 – e-(0.112) (8)) = (1) (1) (e-(0.112) (8)) = 253 or ~ 250 mg Question # 3 A 10-year-old, 32 kg female, is receiving valproic acid sprinkles 250 mg (2 * 125 mg) PO Q 8 hr for her seizure disorder. Calculate her valproic acid level at steady state. Answer: (S) (F) (Dose / t) Cpss = Cl Cl = (13 mL/kg/hr) (32 kg) = 416 mL/hr or 0.416 L/hr (S) (F) (Dose / t) Cpss = Cl (1) (1) (250 mg/8 hr) = 0.416 L/hr = 75.1 mg/L Question # 4 A 23-year-old, 70 kg female, is receiving phenobarbital 60 mg BID and phenytoin 300 mg QD. The steady-state plasma drug concentrations are 18 mg/L for phenobarbital and 10 mg/L for phenytoin. Because of poor seizure control, valproic acid 500 mg Q 8 hr was added to this drug treatment regimen. Two months later, she complained of increased drowsiness. At that time, plasma drug concentrations were measured and reported as follows: phenobarbital 30 mg/L; phenytion 6 mg/L; and valproic acid 75 mg/L. How can the increased phenobarbital concentration and decreased phenytoin concentration be explained? Answer: Valproic acid decreases the clearance of phenobaraital by about 40%. The increase in plasma phenobarbital concentration from 18 mg/L to 30 mg/L 60 days after valproic acid was added is consistent with a 40% decrease in phenobarbital clearance. If the original steady-state phenobarbital concentration of 18 mg/L is desired, the daily phenobarbital dose should be decreased by about 40%. The decline in plasma phenytoin concentration from 10 mg/L to 6 g/L is probably the result of competition for plasma protein binding between valproic acid & phenytoin. In patients with normal serum albumin concentrations & normal renal function, valproic acid (at a concentration of ~ 70 mg/L) produces a 30% - 40% decline in phenytoin plasma concentrations. This acute decline in the plasma concentration of phenytoin is due to a decrease in the bound concentration. The unbound or therapeutically active plasma phenytoin concentration appears to remain unchanged because any bound phenytoin which is displaced re-equilibrates with the large tissue compartment. Since the tissue space is large, the increased tissue concentrations will be negligible. There is less well-documented evidence that chronic valproic acid therapy increases the unbound phenytoin concentrations; this may be caused by inhibition of phenytoin metabolism. To minimize the impact competitive plasma protein binding on the assessment of drug concentrations, both phenytoin & valproic acid samples should be obtained when valproic acid concentrations are at their lowest (that is, at trough, or just before the next dose). Question # 5 "Jane" is an 18-year-old woman with a history of generalized tonic-clonic seizures and myoclonic jerks. Her seizures were poorly controlled with carbamazepine and phenytoin, but she has been seizure-free for 2 years since starting valproic acid. However, over the past year she has gained 20 pounds. Upon review of systems, she also reports irregular menses for the past 6 months. Explain. Answer: Weight gain is a particularly common side effect of valproic acid. One study found that 57% of adult patients gained more than 4 kg during valproic acid therapy. Another study found that 50% of adult patients gained an average of 7 kg without any change in diet or exercise. In a retrospective study of 100 children treated with valproic acid, 44 had significant weight gain. This side effect does not appear to be dose-related, but it can be minimized by diet and exercise. Serum sex hormones and vaginal ultrasonography were studied in 98 women with epilepsy and found menstrual disturbances in 10 (43%) of the 23 women being treated with valproate alone. Six women had polycystic ovaries, and three had elevated serum testosterone. Back