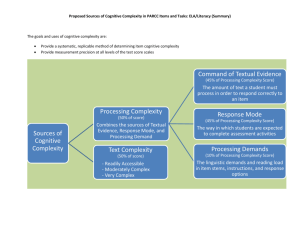
Word Document
advertisement

Karmiloff-Smith & Thomas, page 1 To appear in Special Issue of Development and Psychopathology: “Contributions of Nature to Developmental Theory” What can developmental disorders tell us about the neurocomputational constraints that shape development? The case of Williams syndrome Annette Karmiloff-Smith1 & Michael Thomas2 1 Neurocognitive Development Unit, Institute of Child Health, London 2 Birkbeck College, University of London Karmiloff-Smith & Thomas, page 2 Abstract The uneven cognitive phenotype in the adult outcome of Williams syndrome has led some researchers to make strong claims about the modularity of the brain and the purported genetically determined, innate specification of cognitive modules. Such arguments have particularly been marshalled with respect to language. We challenge this direct generalisation from adult phenotypic outcomes to genetic specification, and consider instead how genetic disorders provide clues to the constraints on plasticity that shape the outcome of development. We specifically examine behavioural studies, brain imaging and computational modelling of language in Williams syndrome, but contend that our theoretical arguments apply equally to other cognitive domains and other developmental disorders. While acknowledging that selective deficits in normal adult patients might justify claims about cognitive modularity, we question whether similar, seemingly selective deficits found in genetic disorders can be used to argue that such cognitive modules are pre-specified in infant brains. Cognitive modules are, in our view, the outcome of development, not its starting point. We note that most work on genetic disorders ignores one vital factor: the actual process of ontogenetic development, and argue that it is vital to view genetic disorders as proceeding under different neurocomputational constraints, not as demonstrations of static modularity. Karmiloff-Smith & Thomas, page 3 I. Introduction At times, genetic mutations give rise to atypical development from embryogenesis onwards. Some are inherited but others are due to purely random events. One such genetic disorder – Williams syndrome (WS) – is caused by the chance misalignment during meiosis of identical flanking regions surrounding some 25 genes on one copy of chromosome 7q11.23 (see Donnai & Karmiloff-Smith, 2000, for review). Because of its uneven cognitive phenotype in the adult outcome, WS has given rise to strong claims about the modularity of the human mind and the purported genetically determined, innate specification of such modules (Pinker, 1994, 1997, 1999; Smith, 1999; Smith & Tsimpli, 1995). But can one generalise directly from adult phenotypic outcomes to genetic specification in this way? In this paper we argue against the simplicity of such claims and consider instead the way in which genetic disorders are informative about the constraints on plasticity that shape the outcome of processes of development. In doing so, we raise a number of crucial questions. First, can one use the selective deficits found in the mature, previously normally developed brain of adult neuropsychological patients to make claims about the cognitive modularity of the human brain? Second, if similar, seemingly selective deficits are also found in genetic disorders, can these be used to argue that such cognitive modules are genetically determined and pre-specified in the infant brain? We will answer in the affirmative to the first question, with a caveat about whether such pure deficits truly exist. But our response will be negative to the second question, arguing that most work on genetic disorders ignores one vital factor: the process of ontogenetic development. In particular, we submit that it is vital to view genetic disorders as proceeding developmentally under different neurocomputational constraints, not as Karmiloff-Smith & Thomas, page 4 demonstrations of static modularity. We will illustrate our arguments with the case of language acquisition in Williams syndrome, although we maintain that the general tenets apply equally to other domains of cognition and to other developmental disorders. II. Selective Deficits in Adult Neuropsychological Patients It is now well established that adults who had previously developed normally can, in the case of focal damage due to stroke or trauma, display selective behavioural deficits (Cappelletti, Butterworth & Kopelman, 2001; Cipolotti, Butterworth & Warrington, 1995; Coltheart, 2002; Patterson, 1981; Rapp & Caramazza, 2002; Shallice, 1988). The existence of such patients has led to the postulation that the brain is composed of modules, each dedicated to a specific kind of input processing. Patients with severe agrammatism may present with other aspects of cognition, including non-grammatical aspects of language, that seem to function normally. Likewise, other patients may have serious word-finding difficulties but their syntactic expression is fluent. This suggests that, by adulthood, specialised functions have become relatively localised to specific brain regions (in this case, that processing of grammar and word-specific knowledge relies on distinct underlying circuitry). However, it is important to recall that such an inference from selective deficit to normal structure is predicated on the assumption that impaired behaviour can be traced to damaged underlying circuitry and that intact behaviour can be traced to residual normally functioning circuitry. This assumption corresponds to the idea that following focal damage, the rest of the system continues to function exactly as it did before the brain insult, thereby giving rise to scores in the normal range. Of course, in attempting to establish that apparently intact functions are indeed working just as they Karmiloff-Smith & Thomas, page 5 did before, the researcher is always at the mercy of the sensitivity of the measurement scale used. Our own work on adults with genetic disorders has demonstrated that even when behavioural scores fall within the normal range on standardised tasks, this does not necessarily entail normal underlying cognitive processes (Grice, Spratling, Karmiloff-Smith, Halit, Csibra, de Haan, & Johnson 2001; Karmiloff-Smith, 1997, 1998; see also the process/achievement distinction introduced by Werner, 1937). Nonetheless, in the case of the previously normal adult patient and to the extent that genuine cases of pure dissociations of behaviour indeed exist, then those data tend to point to relative modularity of mind. III. Selective Deficits in Patients with Genetic Disorders What about selective deficits in adults with genetic disorders? Do they suggest that the mind starts off with independent modules that can be selectively spared or impaired? First, it must be recalled that individuals with genetic disorders have not developed normally to adulthood and then suddenly suffered a brain insult; their brains have developed atypically from the outset. Yet, based on arguments from the adult neuropsychological model, there exist numerous claims in the literature for selective deficits in disorders of a genetic origin, against a background of intact or preserved function, often allied to claims that such selective deficits are evidence that cognitive modules are innate (Baron-Cohen, 1998; Leslie, 1992; Pinker, 1999; Smith, 1999; Smith & Tsimpli, 1995; Temple, 1997). It should be noted that the terminology used here is misleading. While researchers frequently employ terms like “intact” and “preserved” with respect to cognitive functions in genetic disorders, this is not what they actually mean. The terms intact Karmiloff-Smith & Thomas, page 6 and preserved imply that a pre-existing system has not been damaged. This may well be appropriate for adults who have suffered damage to a previously normal cognitive system, but there is no equivalent pre-existing state for the developmental disorder. When researchers deploy terms like “intact”, “preserved”, or “normal” for a genetic disorder, they are using shorthand, and they usually mean two things. First, they mean that behavioural scores on certain standardised tests fall within the normal range. But, second, they are proposing that the underlying cognitive processes have developed normally. The unfortunate prevalence of this terminological shorthand has tended to obscure the fact that claims about intact and impaired functions in genetic disorders actually constitute implicit developmental theories. More importantly, our view is that in developmental disorders, the existence of pure deficits alongside so-called “intact modules” can be challenged not just empirically, but theoretically as well. Even if selective, modular-like deficits were to exist in the older child and adult, we argue that modules are the result of a process of development, not its starting point (Karmiloff-Smith, 1992). The effects of a genetic mutation during embryogenesis and postnatal brain growth are likely to be widespread across the developing system. Some domains will be more affected than others due to the different features of their particular problem space (Karmiloff-Smith, 1998; Karmiloff-Smith, Scerif & Thomas, 2002). It is crucial to take into account how development itself might alter final outcomes. For instance, a tiny impairment in infancy could impact differentially over developmental time, such that many domains are affected but some display only very subtle deficits, whereas others are much more obviously impaired. Some systems may develop atypically but still be able to generate behavioural scores that fall within the normal range on coarse standardized Karmiloff-Smith & Thomas, page 7 psychological tests. Since cognitive systems tend to interact during development (either directly or indirectly via the environment), a cognitive system that is developing atypically may begin to subtly perturb the developmental trajectories of other systems with which it interacts, and particularly those that attempt to compensate for emerging behavioural impairments. The complex dynamics of interaction between neurocomputational constraints, the environment and ontogeny cannot be ignored. IV. Constraints on Plasticity Despite the above caveats about the interpretation of uneven cognitive profiles, it is important to recognise that developmental disorders can actually be very informative about constraints on plasticity. Plasticity is often invoked only in terms of the brain’s response to hemispherectomies after epilepsy, to early or late damage and the like, as if plasticity were a rare developmental process. As Wexler (1996) has put it: "It is uncontroversial that the development [of Universal Grammar] is essentially guided by a biological, genetically determined program....Experience-dependent variation in biological structures or processes...is an exception...and is called 'plasticity' “ Yet in our view, the notion of genetic determinism is misguided; plasticity is the rule, not the exception (see, also, Cicchetti & Tucker, 1994, for a similar argument). Indeed, plasticity is central to all development, normal or atypical, and explains how structure changes as a function of experience. But plasticity is not, of course, unconstrained. The initial properties of a learning system shape how change occurs following experience. The adult brain is in fact the sculpted result of a complex interaction between the Karmiloff-Smith & Thomas, page 8 individual’s processing of a wide variety of structured inputs and the neurocomputational constraints on the developing brain. A crucial goal, therefore, is to identify those constraints. But in identifying relatively consistent brain structures in the normal adult, as in the normal child, it is difficult to distinguish two possible scenarios: either the consistent structures emerge from consistent regularities in the environments to which children are exposed – what some have called “species-typical environments” (Johnson & Morton, 1991) – or they arise from consistent neurocomputational constraints that shape the course of development despite variability in the environment. It is here that experiments of nature may further our endeavours: some children start life with brains that have atypical neurocomputational constraints but they live in relatively normal environments, whereas other children may start out with potentially normal brains but from the outset grow up in very atypical environments (in utero due to drugs or alcohol, or in the outside world due to impoverished or violent conditions). This distinction has also been discussed by Cicchetti (2002). The former case is the focus of the present paper (for the latter – radically atypical environments – see review by Thomas, in press). We submit that genetic developmental disorders can indeed inform theories of normal development provided they are viewed as altered constraints on neural plasticity in a developing organism and not as illustrations of “intact” versus “damaged” static modules. V. Atypical Plasticity As mentioned above, sometimes a specific deficit arises after a focal lesion to the normal adult brain. However, as we shall shortly see, when a normal child suffers the same focal lesions as an adult but in early development, selective deficits rarely ensue. This is because normal processes of plasticity kick in and usually lead to Karmiloff-Smith & Thomas, page 9 recovery from early insult, with no serious, lasting impairments. What about the case of genetic developmental disorders without focal brain damage? Why doesn’t plasticity simply compensate for the genetic mutation and allow the child to recover function in similar ways to normal children with early focal brain damage? We believe this is because of the improbability that the genetic mutation merely affects a single, specific domain; rather, it is likely to affect plasticity itself, because genetic mutations can give rise to atypical neurocomputational constraints from the outset (Karmiloff-Smith, Scerif, & Thomas, 2002). In this article, we use one genetic disorder, Williams syndrome (WS), to illustrate how the atypical developmental pathways followed by this disorder may provide clues to the normal neurocomputational constraints on development. We focus here particularly on the domain of language acquisition, because it is WS language that several authors have embraced as the perfect illustration of an intact grammar module and the example of a dissociation between language and the rest of cognition (Pinker, 1991, 1994, 1999; Smith, 1999). Where appropriate, we contrast WS with normal children who have experienced early brain damage and those suffering from other developmental disorders of genetic origin. However, prior to doing so, we provide an account of the WS genotype and phenotype in order to situate our subsequent arguments. VI. A Specific Example: Williams Syndrome Williams syndrome (WS) is a rare neurodevelopmental disorder occurring in approximately 1 in 20,000 live births (Morris, Demsey, Leonard, Dilts, Blackburn, 1988). It gives rise to specific physical, behavioural and cognitive abnormalities, Karmiloff-Smith & Thomas, page 10 together with structural, chemical and functional anomalies in the developing brain (Bellugi, Wang & Jernigan, 1994; Grice et al., 2001; Mervis, Morris, Bertrand & Robinson, 1999; Rae, Karmiloff-Smith, Lee, Dixon, Grant, Blamire, Thompson, Styles & Radda, 1998). The syndrome was initially reported by cardiologists (Beuren, Apitz & Harmjanz, 1962; Williams, Barratt-Boyes & Lowe, 1961), who discovered an association between the existence of supravalvular aortic stenosis (SVAS) and a characteristic facial dysmorphology, together with growth retardation and learning difficulties. Known as Williams syndrome in the USA and Williams-Beuren syndrome in Continental Europe, it was initially called Idiopathic Infantile Hypercalcaemia in the UK because some infants with a similar clinical description also presented with infantile hypercalcaemia (Black & Bonham-Carter, 1963). However, this turned out not to be a defining characteristic in subsequent cases, and so the disorder is now also referred to as Williams syndrome in the UK. A. The WS genotype Most cases of Williams syndrome are sporadic, although a few instances of concordant monozygotic twins and of parent to child transmission have been reported in the literature (Morris, Thomas & Greenburg, 1993; Pankau, Gosch, Simeoni, & Wessel, 1993). Although for some 20 years WS was diagnosed only on the basis of clinical criteria, the early 1990s hailed the discovery of the first genetic markers of the syndrome (Curran, Atkinson, Ewart, Morris, Leppert, & Keating, 1993; Ewart, Morris, Ensing, Loker, Moore, Leppert, & Keating, 1993a). Curran and collaborators pointed to an association between a disruption due to the translocation of one copy of the elastin gene at chromosome 7q11.23 and the existence of supravalvular aortic stenosis, a common feature of WS. Ewart and colleagues then confirmed that Karmiloff-Smith & Thomas, page 11 hemizygosity at the elastin locus also occurred in patients with WS (Ewart, Morris, Atkinson, Weishan, Sternes, Spallone, Stock, Leppert, & Keating, 1993b). The elastin gene is expressed during the third trimester in utero and during early postnatal life. Its deletion leads to the production of structurally abnormal tropelastin and causes problems with elasticity and connective tissue in numerous parts of the organism, particularly the skin and arteries. The microdeletion measures some 1.5Mb and is of fairly uniform size in the majority of patients with WS, with the elastin gene being midway between the two breakpoints (Perez-Juralo, Peoples, Kaplan, Hamel & Franke, 1996). Parental origin seems to play no role, with deletions on the maternally and paternally inherited chromosomes occurring with equal frequency. The mutational mechanism seems to lay in unequal meiotic recombination between chromosome 7 homologues, although intrachromosomal rearrangements also arise. More recently, it has been shown that large repeats containing genes and pseudogenes flank the two deletion breakpoints, thereby lending themselves to chance misalignment (Peoples, Franke, Wang, PerezJurado, Paperna, Cisco, & Franke, 2000; Robinson, Waslynka, Bernasconi, Wang, Clark, Kotzot, & Schinzel, 1996; Urban, Helms, Fekete, Csiszar, Bonnet, Munnich, Donis-Keller & Boyd, 1996). Once the locus of the WS deletion was documented, several others genes started to be identified in the critical region. Much excitement was generated by the discovery of the deletion of the LIM Kinase-1 gene because it is expressed in the brain. LIMK1 is situated telomeric to ELN and was found to be deleted in all patients with a typical WS deletion (Frangiskakis, Ewart, Morris, Mervis, Bertrand, Robinson, Klein, Karmiloff-Smith & Thomas, page 12 Ensing, Everett, Green, Proschel, Cutowski, Noble, Atkinson, Odelberg & Keating, 1996; Tassabehji, Metcalfe, Fergusson, Carette, Dore, Donnai, Read, Proschel, Gutowski, Mao & Sheer, 1996). LIMK1 encodes a protein tyrosine kinase that inactivates cofilin, a protein required for the turnover of actin filaments (Arber, Barbayannis, Hanser, Schneider, Stanyon, Bernard & Caroni, 1998). Mouse models had already shown that during embryogenesis LIMK1 is expressed in the central nervous system, including the inner nuclear layer of the retina, the cortex, the spinal cord, the cranial nerves and dorsal root ganglia (Proschel, Blouin, Gutowski, Ludwig, & Noble, 1995). Thus, mutations in the expression of LIMK1 in Williams syndrome are likely to affect axonal guidance during the crucial building of the central nervous system (for a review of the genes in the WS critical region, see Franke, 1999). B. The WS physical phenotype Mean birth weight in WS is low, with postnatal growth retardation frequently reported. Early puberty also often contributes to the low final adult height (Cherniske, Sadler, Schwartz, Carpenter & Pober, 1999). The facial dysmorphology is particularly characteristic. Patients have a flat nasal bridge and anteverted nares, a wide mouth with fleshy lips, periorbital fullness, flat malar region, small mandible and prominent cheeks. Failure to thrive, often due to difficulties in sucking, is reported during early infancy. A prematurely aged appearance is often apparent in late adolescence and early adulthood. As mentioned, supravalvular aortic stenosis and peripheral pulmonary artery stenosis are very common in the syndrome (Hallidie-Smith & Karas, 1988), with elevated blood pressure also frequently noted in adolescents and adults (Broder, Reinhardt, Ahern, Lifton, Tamborlane & Pober, 1999). Some 50% of children with WS have strabismus, commonly with refractive errors (Atkinson, Karmiloff-Smith & Thomas, page 13 Anker, Braddick, Nokes, Mason & Braddick, 2001; Winter, Pankau, Amm, Gosch & Wessel, 1996). Hyeracusis – an unusual hypersensitivity to certain sounds – is present in as many as 95% of patients, whereas audiometry is usually normal. Little is yet known about central auditory processing in Williams syndrome although new research is underway (Cohen, Ansari, Rosen & Karmiloff-Smith, 2002). C. The WS behavioural and cognitive phenotypes In experimental psychology and cognitive neuroscience, the terms “behavioural” and “cognitive” have different meanings from the way in which they are employed in clinical medicine. For the clinician, “behavioural phenotype” refers to emotional and personality traits, attention deficits, as well as IQ scores. “Cognitive” is simply subsumed under the term “behavioural” and refers to the standardised measures of intelligence. In experimental psychology, by contrast, “behavioural” refers to measures of overt behaviour, e.g., scores relating to whether or not the subject succeeds or fails at a task. The “cognitive” level attempts to account for the mental processes underlying the overt behaviour, i.e., how the individual processes the inputs involved in the task. This is not unsimilar to an early but hitherto rather neglected distinction made by Werner (1937) between achievement and process. It must be always recalled, therefore, that the same overt behaviour may be achieved by different underlying cognitive processes when comparing various disorders to the normal case (Karmiloff-Smith, 1998). Thus, claims that certain aspects of a profile are “unimpaired”, “intact”, “preserved” and the like, need to be taken with caution until the cognitive level has been fully explored. To understand a syndrome in any depth, it is vital to distinguish between the behavioural phenotype and the cognitive phenotype. Karmiloff-Smith & Thomas, page 14 The behavioural phenotype in WS includes what has been termed ‘hypersociability’ (Jones, Bellugi, Lai, Chiles, Reilly, Lincoln & Adolphs, 2000), with a tendency to be overly friendly with strangers and to lack social judgement skills (Einfeld, Tonge & Florio, 1997; Gosch & Pankau, 1997). People with WS frequently display high anxiety about new situations. They are, however, particularly empathetic towards others’ emotions, but far less skilled at understanding human intentionality (TagerFlusberg, Boshart & Baron-Cohen, 1998). Most studies estimate a mean full IQ of between 51-70 (Bellugi, et al., 1994; Mervis et al., 1999; Udwin & Yule, 1991). To be noted, however, is the fact that full IQ scores camouflage marked unevenness in the WS profile, with verbal IQ outstripping performance IQ in the majority of cases. The pioneering work of Bellugi and her collaborators had originally suggested some clearcut dissociations in the cognitive architecture of WS. Language and face processing appeared to be “preserved” alongside both general retardation and particularly serious problems with visuo-spatial cognition (Bellugi, et al., 1994). However, as mentioned above, the notion that behavioural abilities in a developmental disorder directly index underlying cognitive processes is one that we challenge (Karmiloff-Smith, 1998; Thomas & Karmiloff-Smith, in press, a). Indeed, in-depth analyses of the language and face processing of WS adults – two areas frequently reported to be “intact”– strongly suggest that the behavioural proficiencies of these individuals are supported by different cognitive processes compared with normal controls. Karmiloff-Smith & Thomas, page 15 VI. Neurocomputational constraints on development in Williams syndrome Having set the stage for understanding the characteristics of our particular experiment of nature – Williams syndrome – we can now turn to the issues laid out in our introductory sections as to what developmental disorders can tell us about the neurocomputational constraints that shape development, using language acquisition as our main illustration. The rest of this article will proceed as follows. We begin with a consideration of the (static) endstate of language development in WS, including claims for selective deficits and dissociations. To emphasise the role that alteredconstraints-on-plasticity must play in WS, we identify the type of acquired damage that causes similar patterns of behavioural deficits in normal adults, and then examine what happens to normal children who experience such damage early in development. As we have seen, in our target disorder the cause is not acquired damage but a genetic mutation acting from embryogenesis onwards. Consideration of a realistic causal link between (potentially mutated) genes and (potentially altered) behaviour leads us to focus on the precursors of language development in WS. It is here that we have the best opportunity to reveal the intrinsic neurocomputational constraints that differ in the infant with WS, before they are obscured behind layers of development driven by dynamic interaction with the environment. A review of the available evidence leads us to a hypothesis concerning the constraints that may be atypical in WS language (and by implication, that must act appropriately in normal development). Armed with these, we offer a developmental theory of the acquisition of language in this disorder and how this might enlighten theories of typical development. Finally, we focus on how one might explore more precisely the influence of constraints on development Karmiloff-Smith & Thomas, page 16 within a neurocomputational framework via the application of connectionist modelling techniques. A. Language in late childhood/adulthood in Williams syndrome In the initial characterisations of Williams syndrome, it was thought that the seeming dissociation between language and other general reasoning abilities might represent an informative ‘experiment of nature’. Thus, Rossen, Klima, Bellugi, Bihrle & Jones (1996) commented that the syndrome ‘presents a remarkable juxtaposition of impaired and intact mental capacities…[..]...linguistic functioning is preserved in Williams syndrome while problem solving ability and visuospatial cognition are impaired’. In Pinker’s initial references to the disorder (1991, 1994), he viewed WS as an example of the potential developmental independence of language and cognition, consistent with his theory that language (and particularly syntax) is an innate, selfcontained module. Indeed, he contrasted WS with Specific Language Impairment (SLI), a disorder that presents as a case of impaired language development with apparently normal non-verbal cognition and intelligence. Given that SLI has a heritable component, Pinker (1999) argued that WS and SLI together represent a ‘genetic double dissociation’ pointing to the developmental independence of language from cognition. However, subsequent careful research has led researchers to more refined claims, given that a range of studies has revealed anomalies at all levels of language in WS, including phonology, lexical-semantics, grammar, and pragmatics. Such studies include the examination of WS populations in several countries, such as the USA, Britain, Italy, France, Spain, and Germany (see Thomas & Karmiloff-Smith, in press, Karmiloff-Smith & Thomas, page 17 a, for a review of this work). It does appear true that in some cases, individuals with WS are competent at understanding and producing a wide range of sophisticated grammatical constructions (see, e.g., Clahsen & Almazan, 1998). (It should of course be kept in mind that, as in many developmental disorders, there is great individual variability in WS. Some lower functioning individuals show much poorer language despite having the same genetic deletion.) On the other hand, in almost all cases of WS, language performance falls below that found in chronological age-matched controls. Indeed, most studies of language in WS now compare performance against mental age controls. Paradoxically, those claiming that language and cognition are independent also opt for MA-matched controls; yet this very choice implicitly accepts a relationship between language and cognition! And it is worth noting that full IQ measures make the language results look more impressive than they really are. To state that a 25-year-old adult with WS has a full IQ of 53 but understands complex embedded clauses seems impressive. But assessed from a different angle, that of Mental Age, it is much less amazing. The same 25-year-old individual with WS may have a Verbal Mental Age of 8, but then there is nothing surprising about the fact that typically developing 8-year-olds have mastered many complex, structural aspects of language (see discussion in Karmiloff-Smith, 1998). It is often the pragmatic limitations of WS language that draw listeners’ attention to the fact that this individual’s conversation is not at the level we expect for a typical 25-year-old, or an 8-year-old, for that matter. For example, in WS, speech content is often odd or out of place in a particular social context (Volterra, Capirci & Caselli, 2001), speech can contain high levels of clichés and stereotyped phrases (Howlin, Davies & Udwin, 1998a), and the comprehending of non-literal language, can be very deficient (Howlin, Davies & Udwin, 1998b). Karmiloff-Smith & Thomas, page 18 Despite a retreat from some of the stronger claims, two aspects of language in WS remain notable. First, the language of these individuals is often more advanced than that found in other genetic developmental disorders with comparable IQ, such as Down’s syndrome (Bellugi, Wang & Jernigan, 1994; Mervis et al., 1999). Second, language performance in WS is markedly out of step with their visuo-spatial cognition, which is particularly poor and shows a characteristic ‘featural’ style of processing (Deruelle, Mancini, Livet, Cassé-Perrot, & de Schonen, 1999; Donnai & Karmiloff-Smith, 2000). Indeed, one of the defining criteria of the cognitive phenotype of WS is the disparity between performance on standardised tests of vocabulary and on standardised tests of visuo-spatial construction (such as a copying a pattern with a set of coloured blocks, or copying a picture) (Mervis, Robinson, Bertrand, Morris, Klein-Tasman, & Armstrong, 2000). However, claims persist that selective deficits can be found within the language systems of individuals with WS, and that such deficits can inform theorists about the structure of the normal language system. For instance, Temple and colleagues argue that ‘the linguistic performance of [individuals with] WS can be explained in terms of selective deficits to an otherwise normal modular system’ (Temple & Clahsen, in press, italics added), and as a pattern of ‘some preservation and some disability’ (Temple, Almazan, & Sherwood, 2002). The latter claims for deficits have focused on the representation of and access to word-specific knowledge. For instance, the language of individuals with WS has frequently been characterised as containing unusual or low frequency items (e.g., Bellugi et al., 1994; Pinker, 1991, 1994, 1999; Rossen et al., 1996; Temple et al., 2002), and older children with WS have been Karmiloff-Smith & Thomas, page 19 argued to demonstrate naming deficits and difficulties representing fine semantic distinctions (Temple et al., 2002). In the domain of inflectional morphology, it has been argued that word inflections involving word-specific knowledge (such as irregular past tenses and plurals) are selectively impaired in WS, while those following grammatical rules are in line with mental age (Clahsen & Almazan, 1998, 2001; Pinker, 1991, 1994), but, note, not chronological age. Overall the claim has been that grammar is preserved in WS (i.e., develops normally), while some aspects of word-knowledge are impaired (i.e., develop atypically). To reiterate, the comparison is with mental age, not chronological age, so such claims simply dismiss the considerable delay as if delay in one system were completely independent of the rest of the developing system. WS has none the less been used to support the position that there is a dissociation between language and cognition and that the distinction between grammar and the lexicon is innate (e.g., Pinker, 1999). There are a number of shortcomings with these arguments, not least the fact that many of the empirical findings on which they are based have employed very small numbers of participants with WS and that they have often failed to replicate when larger samples have been used (e.g., standardised tests of word fluency reveal that WS vocabulary is no more atypical than mental age controls: Jarrold, Hartley, Phillips & Baddeley, 2000; Scott, Mervis, Bertrand, Klein, Armstrong, & Ford, 1995; there is no significant selective deficit for irregular inflections over regular inflections: Thomas, Grant, Gsödl, Laing, Barham, Lakusta, Tyler, Grice, Paterson & Karmiloff-Smith, 2001, and Zukowski, 2001; naming skills are in line with verbal mental age: Thomas, Dockrell, Messer, Parmigiani, Ansari & Karmiloff-Smith, 2002). However, the selective-deficit claims are illustrative of the view that it is sufficient to explain a Karmiloff-Smith & Thomas, page 20 developmentally disordered system with reference to a static model of the normal adult language system, and to orient an empirical program merely towards identifying those components that are deficient – thereby negating altogether the contribution of development. We argue that this appeal to static models has inhibited progress in understanding WS in particular and developmental disorders in general, but we do not wish to overstate this case. Even in the more static accounts, with their postulation of preserved and impaired components, there are occasional hints among researchers that the final explanation of anomalies in the WS system must fall within a developmental framework. Thus, Pinker comments that ‘presumably LIM-kinase1 [deleted from one copy of chromosome 7 in WS] plays an important role in the development of the neural networks used in spatial reasoning, possibly in the parietal lobes. The other missing genes, perhaps, are necessary for the development of other parts and processes of the brain, though not for language’ (1999, p.260-1, italics added). When Rossen et al. (1996) appealed to a static model of the WS lexicon to explain the presence of unusual words in WS vocabulary, they sought to characterise the problem as a specific anomaly of activation dynamics against a background of normal lexical structure: ‘… While individuals with WS have well organised semantic categories, and have good access to word knowledge, an anomaly does exist in some tasks dependent on consideration of words in a complex activation environment’. Yet in the same article, when these researchers began to consider the impact of development on the disorder, they added that, so long as activation dynamics were involved in knowledge acquisition, knowledge could not be normal while activation dynamics were anomalous: ‘Contemporary neural models of learning postulate definable Karmiloff-Smith & Thomas, page 21 quantitative relationships bridging the classical dichotomy of structure and process… Within this model paradigm, if activity malfunction with the lexicon exists, then abnormality of lexicon structure will likely follow’ (Rossen et al., 1996). Nevertheless, the prevalence of theories of the “preserved/impaired” kind provides an opportunity to stress our alternative claim that genetic developmental disorders are best viewed as altered constraints on plasticity. To make this argument, we first consider what type of brain damage might underlie the types of selective deficits proposed, were they to appear in a normal adult. We then turn to the literature on the developmental outcomes of these types of brain damage when they occur in typically developing young children, in whom we assume that normal constraints on plasticity hold. The clearest parallels between behavioural deficits in WS and those found in normal adults with acquired brain damage have been made in the domain of word inflections (e.g., past tense of verbs, plurals of nouns, comparatives of adjectives). Thus Pinker (1991, 1994) has explicitly argued that the deficits in inflectional morphology found in WS are similar to those occurring in fluent aphasia in normal adults, following damage to left temporal areas. These patients also show difficulties in word retrieval. Similar parallels are drawn with deficits displayed in neurodegenerative disorders, such as Alzheimer’s disease and cases of semantic dementia. These disorders are also characterised by acquired functional damage that is greater in the temporal areas and adjacent parietal areas than in the frontal lobes. And for reference, as the earlier quoation from Pinker implies, visuo-spatial constructive deficits are found in normal Karmiloff-Smith & Thomas, page 22 adults following damage to parietal areas, and in particular, the featural style of processing tends to be associated with right parietal damage. Note, however, that individuals with WS do not suddenly suffer discrete brain damage in adulthood, but have anomalies in brain structures from the start of brain development. What happens if these types of focal brain damage – to left temporal areas, or to right parietal areas – occur to normal individuals early in infancy? Another set of nature’s experiments can provide the answer. B. Evidence for atypical constraints on plasticity in Williams syndrome Bates and Roe (2001) recently reviewed the evidence concerning the effects of early unilateral brain damage on the language development of young children, including the effect of lesion side (left or right hemisphere) and lesion site. There were three main findings. First, almost all the brain injured children exhibited delays in first-word production, regardless of lesion side or site. Bates and Roe interpret these data as suggesting that it is hard to get language “off the ground” after significant damage to either hemisphere. Second, delays in word production tended to be more severe in children with left posterior damage, particularly in the temporal lobe. Bates and Roe note the apparently greater importance of the left hemisphere, but interestingly the fact that in the adult, it is left frontal not temporal posterior damage that is associated with expressive language deficits. Third, and most relevant to our concerns, is the fact that the evidence suggests that when damage occurs prior to around 5 to 7 years of age, plastic reorganisation takes place such that when tested later, these children show little if any language impairment. Importantly, there are no broad effects of side of Karmiloff-Smith & Thomas, page 23 damage. In short, early damage to left (or right) hemisphere when there is normal plasticity tends to lead to recovery, with no long-lasting gross deficits. The implication is that the analogies drawn between behavioural deficits after focal lesions in normal adults and behavioural deficits in individuals with WS do not point to a common underlying cause. Where focal damage occurs in early childhood, normal plasticity is sufficient to effect behavioural recovery as a result of ontogenetic development. If we rule out focal damage + normal plasticity as an explanation for WS, then several options remain. There could be widespread damage + normal plasticity, focal damage + atypical plasticity, or widespread damage + atypical plasticity. Of these options, increasing evidence suggests that the brains of individuals with WS do not present with focal lesions (and this is also the case for almost all other genetic developmental disorders), but with widespread differences in neuroanatomical features (Bellugi, Wang & Jernigan, 1994), in neuronal density (Galaburda, Wang, Bellugi & Rossen, 1994), in biochemistry (Rae et al., 1998), and in electrophysiological activity (Grice et al., 2001; Neville, Mills & Bellugi, 1994). The possibility that WS is caused by widespread but subtle damage + normal plasticity merits consideration. Rourke (e.g., 1987, 1989) proposed the notion of nonverbal learning disability to account for a consistent pattern of behavioural deficits observed in children with a history of early, generalised cerebral deficits. Anderson, Northam, Hendy and Wrennall (2001) summarise the main features as follows: bilateral tactile-perceptual deficits, more marked on the left side of the body, impaired visual recognition and discrimination, impaired visuospatial organisation, bilateral psychomotor coordination problems, again more marked on the left side of the body, Karmiloff-Smith & Thomas, page 24 and difficulties managing novel information. Among these weaknesses are also areas of relative strength, which include simple motor skills, auditory perception, rote learning, selective and sustained attention for auditory-verbal information, basic expressive and receptive language, and word reading and spelling. Non-verbal learning disorder bears a number of similarities to the features of WS, in particular the relative strength of language compared to the deficits in visuospatial skills. Indeed, within language itself, Rourke and Tsatsanis (1996) point to a dissociation between better performance on structural aspects of language than on pragmatics, another similarity with WS. However, differential perceptual and motor problems with the left side of the body have never hitherto been reported in WS. Rourke’s notion of non-verbal learning disability (NVLD) is interesting here in that it is allied to an underlying neurological explanation called the ‘white matter hypothesis’ (Rourke, 1987; see Anderson et al., 2001, for discussion). The idea is that the global connectivity of the brain, the white matter, has a prolonged period of development, in which it is particularly vulnerable to disruption after widespread damage in children who have experienced conditions such as traumatic brain injury, cranial irradiation to treat tumours, or hydrocephalus. (Note that under some circumstances, these conditions may be associated with a normal genotype, such that one might expect normal plasticity if the mechanisms that support it have survived.) Rourke then distinguishes between the roles of white matter in the left and right hemispheres. In the right hemisphere, he proposes that white matter is important for both development and maintenance of skills (including global integration of information), while in the left hemisphere it is important for the development of skills but not necessarily their maintenance. In this view, the right hemisphere is responsible Karmiloff-Smith & Thomas, page 25 for more integrative functions and the left hemisphere for more segregated functions. Widespread damage to the white matter then results in greater impairments to the functions of the right hemisphere (such as visuo-spatial integration and pragmatic aspects of language), than to the more self-contained functions of the left hemisphere, (such as processing of visual detail, auditory processing, basic expressive and productive language). It is an appealing notion that that widespread damage can itself result in differential (seemingly selective) effects on the functions of left and right hemisphere, which normal plasticity is unable to overcome during atypical development (due to the severity of the damage), for this would explain the subsequent uneven profile of cognitive abilities. However, it is far from clear than NVLD is a homogeneous syndrome – indeed, Rourke (1987) prefers to conceptualise it as a ‘final common pathway’ of deficits that may be caused by a range of underlying pathologies. A range of disorders, including both those of genetic origin and of acquired early damage, are subsumed under this heading, each of which shares different levels of similarity to the prototypical description of the disorder. As such, NVLD may be a label attached to the common behavioural deficits shared by very sub-optimal cognitive systems, systems which differ as to their underlying causes and which, at a detailed level, differ in their cognitive symptoms. Gross behavioural deficits in the more challenging, integrative aspects of cognition may then be reached either by a system with normal plasticity but widespread damage (if indeed the mechanisms of normal plasticity remain after such damage), or by a system with both widespread anomalies and atypical plasticity. It is the latter possibility that we would argue obtains in the case of genetic developmental disorders such as WS, for reasons that we address in the next Karmiloff-Smith & Thomas, page 26 section. Nevertheless, despite the common tendency to seek dissociations, the behavioural similarities across genetic syndromes can also be very informative (Bishop, 1997; Karmiloff-Smith, 1997, 1998). Given the complex and indirect relationship between genes and brain development, it remains possible that different genes have similar distal effects on brain development and lead to similar cognitive deficits. Indeed, such a possibility has been explicitly raised regarding similarities identified between WS and velocardiofacial / DiGeorge syndrome (Beardon, Wang, & Simon, 2002), although this initial comparison has yet to be established in depth. C. A causal model for exploring the relationship between genes and behaviour We have argued that the effects of a genetic mutation that emerge during embryogenesis and postnatal brain growth are likely to be widespread across the developing system, albeit relatively subtle, but that a consequence of the mutation can be the emergence of an uneven cognitive profile in the endstate. In trying to link the genetic mutation to the final behavioural profile, it is important to have a realistic causal model in mind that could, however indirectly, connect the two. Despite the fact that in recent times (and often, admittedly, in the popular press), genes have been specifically linked to behaviours (such as the “gene for language”, the “gene for crime”, the “gene for homosexuality”, and so forth), the link between genes and adult behaviour is of course incredibly indirect and involves many-to-many rather than one-to-one mappings between genes and behavioural phenotypes. Moreover, genetic mutations are limited in the ways in which they can disrupt brain development. Pennington (2001) suggests three broad classes of effects, those on (1) Karmiloff-Smith & Thomas, page 27 brain size, in terms of altering the number of neurons or synapses, (2) neuronal migration, sometimes in a regionally specific fashion, and (3) neurotransmission, either by changing levels of neurotransmitter or changing the binding properties of receptor proteins. To this we would add the fact that genetic mutations can also affect the timing of gene expression which will have cascading effects on the developing organism, since timing is a crucial aspect of the emergent organisation of the functional structure (Elman, Bates, Johnson, Karmiloff-Smith, Parisi & Plunkett, 1996; Huttenlocher, 2002; Karmiloff-Smith, 1992, 1998). Importantly, these effects impact on the neurocomputational properties of the brain, including the way in which it modifies its structure as a consequence of internally- or externally-generated activity. Subsequent behavioural deficits are the outcome of a long trajectory of development, whereby cognitive structures emerge via an interaction between internal neurocomputational constraints and the environment. In placing development itself as a vital factor in the explanation of developmental disorders (Karmiloff-Smith, 1998), we have characterised the developmental emergence of cognitive structures as a type of recursive equation (Thomas & Karmiloff-Smith, in press, a). The equation is as follows: More complex cognitive structure = Less complex structure X process of development. This means that the process of development corresponds to the interaction of an internal or external environment with the existing neurocomputational constraints1. In atypical development, the neurocomputational constraints can differ. Under these conditions, each cycle of recursion that operates on a system may produce functional properties that fall inside or outside the normal range, and for any subsequent steps of Karmiloff-Smith & Thomas, page 28 development that rely on this system, a new set of constraints that are more or less atypical for the next pass through the equation. The result is that across the whole developing cognitive system, there emerges a complex, graded pattern of areas that function either within or outside the range of variation that one might expect in the normal population. (In cognitive terms, these graded variations are with respect to the functional properties of sub-systems rather than their purely anatomical or physiological properties). Whether a given subsystem falls inside or outside the normal functional range as a consequence of the operation of the equation depends on the extent to which the development of that subsystem relies upon the particular neurocomputational constraints that were the atypical outcome of the previous operation of the equation2. Ultimately, this cascade of equations emanates from the gene products that were altered in embryogenesis by the original genetic mutation. This is a complex formulation, but we believe it is a more realistic causal pathway than that offered by static theories calling on preserved/impaired modules, because it includes development at every stage. Importantly, as a causal model, it seeks to remain at a single (in this case, cognitive) level of description. Causal relations cannot, in our view, operate across levels of description. That is, neuronal activations do not cause mental representations, they are mental representations. Genes cause molecular events. Thus genes are not only separated from behaviour by a long and complex developmental trajectory, but also by existing at a different, lower level of description. Having taken this point on board, we are presented with a problem. If, to identify the causal origins of cognitive deficits, we must remain at the cognitive level of Karmiloff-Smith & Thomas, page 29 description, then we are faced with the difficulty that the foetus and newborn infant do not have cognition in the way that children or adults do. To construct the causal account, we must therefore describe a proto-cognitive system in the newborn or infant that produces behavioural precursors to later complex behaviours that we see in children and adults (Karmiloff-Smith, 1998). With development, mental representations begin at a simple, concrete level and achieve an increase in abstractness and complexity following operation of the recursive equation we have proposed. Now, some might describe more complex/abstract cognitive processes as “higher-level” functions, and in this sense, the equation recursively climbs from lower to higher levels of representation across development. But crucially, the distinction between low-level and high-level representations is one of complexity and abstractness, while the causal relations remain at the same cognitive level of description. (Of course, different disciplines will seek to characterise this recursive causal equation at other levels of description, such as at the level of neural structure when characterising the emergence of atypical brain structures). We have taken a short diversion from WS to make this conceptual clarification, but the outcome is a clear message of methodology. If we are to tease apart the cascade of developmental equations that has led to cognitive deficits in the adult state, we must pursue those deficits back down the cascade, to their origins in infancy (Brown, Johnson, Paterson, Gilmore, Longhi, & Karmiloff-Smith, 2003; Karmiloff-Smith, 1998; Paterson, Brown, Gsödl, Johnson & Karmiloff-Smith, 1999). In studying cognitive precursors of complex behaviours, we will find the clearest evidence of the altered neurocomputational constraints that are the first cognitive consequence of a genetic mutation. Karmiloff-Smith & Thomas, page 30 D. Atypical precursors to language in Williams syndrome The most salient aspect of the onset of language in WS is that it is delayed. Although this delay is variable, one study of 54 children with WS found an average delay of 2 years, similar to that found for children with Down syndrome (DS) (Singer Harris, Bellugi, Bates, Jones & Rossen, 1997; see also Paterson et al., 1999). Though delayed, some aspects of early development reveal normal behavioural patterns. For example, the onset of hand banging predicts the onset of canonical babbling in infants with WS in the same way as it does in typically developing infants (Masataka, 2001; Mervis & Bertrand, 1997). And, once language development commences, the relationship between vocabulary size and grammatical complexity, although seriously delayed, is again within the normal range, a pattern that contrasts with DS where grammatical complexity is reduced for a given vocabulary level (Singer Harris et al., 1997). Despite the fact that phonological memory appears as a relative strength in WS in childhood and adulthood (Mervis et al., 1999), a study of the ability of infants and toddlers with WS to segment fluent speech stream into words revealed serious delays (Nazzi, Paterson, & Karmiloff-Smith, 2003). In part, then, language delays may be due to problems with the early development of speech perception and phonological representations. However, some precursors appear not just delayed, but atypical. For example, Laing and colleagues examined socio-interactive precursors to language development in toddlers with WS compared with MA controls (Laing, Butterworth, Ansari, Gsödl, Karmiloff-Smith & Thomas, page 31 Longhi, Panagiotaki, Paterson & Karmiloff-Smith, 2002). Although toddlers with WS were proficient at dyadic interactions with a caregiver (and indeed sometimes exceeded the scores of MA controls due to persistent fixation on the caregiver’s face; see also Bertrand, Mervis, Rice & Adamson, 1993; Jones et al., 2000), there was a marked deficiency in triadic interactions incorporating an object. Specifically, toddlers with WS had difficulty switching attention from the caregiver to an object that was being referred to in communication (via pointing, looking, and naming). One might imagine that this deficiency would disadvantage the toddlers with WS in learning the names of objects, since shared attention to newly named objects appears one of the main routes into vocabulary acquisition. And indeed, there is accumulating evidence that precursors to vocabulary development in WS are atypical. Typically developing infants use the presence of linguistic or gestural information accompanying the introduction of novel objects to influence their subsequent categorisation of those objects, sometimes over and above the perceptual similarities among the objects. However, Nazzi and Karmiloff-Smith (2002) found that 2-6 year old children with WS were significantly less able than typical controls to use verbal cues to constrain categorisation. Masataka (2000) found a similar poverty in the ability of 2-3 year olds with WS to use gestural information to constrain categorisation. In typically developing children, the ability to use pointing to refer to objects tends to emerge before the use of verbal labels for the same purpose. Presumably, pointing indexes the emergence of the cognitive ability to make reference, prior to the lexical manifestation. Pointing to objects and eliciting pointing behaviour in adults also Karmiloff-Smith & Thomas, page 32 facilitate the ability to find the correct referent for a given label. However, in WS, Mervis and Bertrand (1997) found that the order was reversed, with the onset of productive vocabulary preceding pointing. Laing et al. (2002) confirmed a deficit in the pointing behaviour of infants with WS, despite relative proficiency at fine motor skills. Vocabulary acquisition, therefore, appears to rely on a different set of cues and constraints in WS. When Stevens and Karmiloff-Smith (1997) examined the constraints that older children and young adults with WS were using to learn labels for novel words, these, too, appeared atypical. It is often the case that WS seems to present with a mixture of typical and atypical patterns within the same domain. For example, Mervis and Bertrand (1997) found that when playing with toys, non-verbal play patterns and object label comprehension patterns in children with WS showed the normal priority of basic level categories over subordinate categories, a pattern also found in DS. However, relations between markers of semantic knowledge and productive vocabulary appeared yet again atypical in the WS group. For instance, spontaneous exhaustive sorting of objects (such as arranging toy animals and blocks into their separate categories) indexes the development of semantic knowledge, and tends to precede a rapid rise in the rate of vocabulary acquisition in typically developing children. Thus, by the time children find it clear which categories objects fall into, it becomes increasingly easier for them to attach consistent labels to different objects. However, for children with WS, Mervis and Bertrand (1997) found no evidence that exhaustive sorting preceded the vocabulary spurt; indeed, several children with WS exhibited the reverse pattern (unlike children with DS, who displayed the normal pattern). Karmiloff-Smith & Thomas, page 33 Finally, anecdotal parental reports have suggested that young children with WS sometimes appear to say more than they actually comprehend (Singer Harris et al., 1997). Consistent with this, there is some systematic evidence that compared to normal children, the vocabulary of young children with WS exhibits a reduced advantage for comprehension vocabulary over production vocabulary (Paterson, 2000), implying a relatively higher productive vocabulary for their level for comprehension. In summary, precursors to language development in WS paint a picture with two main themes. First there is an overall delay, perhaps of a more generalised nature incorporating delays in at least motor, phonological, and semantic development. Second, when language development gets underway, a differential balance emerges between the ability to encode and produce word forms on the one hand, and the acquisition of the semantic underpinnings for those words on the other. E. A developmental framework for language acquisition in Williams syndrome The psychological literature on the language of older children, adolescents, and adults with WS is more substantial, but two types of hypothesis can be identified within it (Thomas & Karmiloff-Smith, in press, a). The first hypothesis is a conservative one, that the language of individuals with WS is broadly in line with their overall learning disabilities. From this perspective, some anomalies arise as an indirect effect of their other deficits, such as the visuo-spatial deficit which causes a difficulty with acquiring prepositions that encode spatial relations (Jarrold, Phillips, Baddeley, Grant & Karmiloff-Smith, 2001). Interestingly, the data also point to a general problem in WS Karmiloff-Smith & Thomas, page 34 with relational terms like “darker than” that are non-spatial and yet involve the need to generate spatialized internal mental representations to process them. However, although this may explain local difficulties in some aspects of WS language, such a conservative hypothesis fails of course to account for why language development should be better in WS than in other disorders with comparable IQ, such as DS. The second hypothesis is the more prevalent and, in line with the conclusions of the preceding section, views language in WS as evolving according to an atypical balance of constraints from phonology and semantics. As yet, there is no consensus on whether the differential balance is caused simply by a relative strength in phonology, simply by a relative weakness in semantics, by a difficulty in integrating these two sources of information, or some combination of these possibilities. Nevertheless, there is now sufficient evidence to begin to sketch out a developmental theory of language acquisition in WS, a theory that stands in marked contrast to accounts that have dealt purely in terms of selective deficits to a static model of the normal system. We have constructed this preliminary account in the context of our recent work exploring the causes of the unusual, socially-engaging vocabulary in the language of individuals with WS (Thomas et al., 2002). This phenomenon is one of its most widely reported characteristics, and a finding that led to the claims about isolated deficits in wordspecific knowledge that we discussed in a previous section. In our preliminary account, language in WS is seen in a wider socio-communicative context, and in a developmental framework that takes into consideration the way in which compensation can occur in a system that has different initial processing biases but can be adapted to meet the social needs of the individual. From this perspective, the unusual Karmiloff-Smith & Thomas, page 35 characteristics of conversation in WS reflect a form of developmental compensation in which language is used primarily to meet the (possibly heightened) social needs of the individual (Jones et al., 2000), but acquisition is restricted by a difficulty in extracting the exact sense and context in which vocabulary items are being used by speaker. Indeed, the final semantic and conceptual representations formed in individuals with WS may well be shallower, with less abstract information and more perceptually-based detail, suggested by work examining conceptual knowledge in WS (Johnson & Carey, 1998), as well as by work on the development of semantic categories and metaphor comprehension (Thomas, van Duuren, Ansari, Parmigiani, & Karmiloff-Smith, 2002). However, language development in WS is bolstered by a relative strength in phonological processing that permits the encoding and production of a range of words. This form of compensation is evidenced indirectly by basic measures of nonword repetition (Mervis et al., 1999), but also directly by analyses suggesting that the contribution of phonology to word learning compared to that of the existing lexicon may be greater in WS than is typical after 5 years of age (Grant, Karmiloff-Smith, Gathercole, Paterson, Howlin, Davies & Udwin, 1997), and from word learning tasks suggesting preferential use of phonological over semantic information (Laing, Hulme, Grant, & Karmiloff-Smith, 2001). The outcome in WS is a language system that is preferentially geared towards engaging and maintaining social interaction, and that uses well-formed socially effective vocabulary and phrases with only approximate semantic underpinnings. Unusual vocabulary in WS is successful in engaging interest, but closer inspection suggests that it is not supported by appropriate contextual nuances of meaning (Rossen et al., 1996). In our view, this sketch emphasises paradigmatic characteristics of the way in which to explore the origin of cognitive deficits in developmental disorders: explore developmental Karmiloff-Smith & Thomas, page 36 precursors in infancy, build these into a dynamic account which incorporates both the social environment of the individual and the capacity for compensation, given the altered constraints acting on development. Against this type of account, we have contrasted an approach to language in WS that only references a static model of the normal system and claims a dissociation between ‘preserved’ syntactic knowledge and ‘impaired’ wordspecific knowledge (Clashen & Almazan, 1998). In fact, we believe that currently there is no strong support for a grammar-lexicon dissociation in WS, nor indeed a theoretical consensus amongst linguists or psychologists that this type of dissociation would be developmentally plausible, given the dynamic and constant interactions between these two aspects of language during normal acquisition. Thus, while processing in the WS lexicon appears inefficient, so too does the processing of syntax in WS (as evidenced by exaggerated patterns of difficulty in WS in sentence processing tasks; see Grant, Valian, & Karmiloff-Smith, 2002; Zukowski, 2001; and Thomas & Karmiloff-Smith, in press a, Thomas et al., 2001, for discussion). The most salient and agreed upon dissociation within the WS cognitive system remains the one first identified by Bellugi and colleagues, of an imbalance between some aspects of language (such as vocabulary) and visuo-spatial processing (see WS Cognitive Profile: Mervis, et al., 1999). And even here, evidence of impairments in spatial vocabulary and spatial representations in WS (Jarrold et al., 2001) suggests that this dissociation is not static but has implications when spatial and language systems interact across developmental time. F. Computational approaches If links are to be made between genotype and phenotype in developmental disorders, we have argued that researchers must identify atypical neurocomputational constraints on plasticity that shape from infancy onwards subsequent trajectories of cognitive Karmiloff-Smith & Thomas, page 37 development. The preceding two sections have presented psychological evidence pointing towards the types of constraints that may be atypical in WS language development. However, notions like phonology and semantics are some way from the neurocomputational characteristics that may have been altered by early neurobiological events and subsequent developmental cascades from embryogenesis onwards. In order to build links between the psychological cognitive level of description and the neural level, it is essential, in our view, to explore the intermediate level of computational learning systems (Karmiloff-Smith, 1992). Such learning systems permit us to build models that encode information at the conceptual level (e.g., word forms and meanings) – thereby making contact with the psychological data – but also for those classes of models that share neurocomputational principles with the underlying substrate, to make contact with the neural level as well. Connectionist networks are an example of this type of modelling and particularly well suited to exploring developmental questions (Elman et al., 1996; Karmiloff-Smith, 1992). We would argue that without computational modelling to link the cognitive and neural levels, researchers are left without a concrete idea of what a ‘developmental process’ might constitute, and what implications it might have in producing or recovering from deficits. It is one thing to stress the importance of development in understanding developmental disorders. It is another to begin to address precisely what the developmental process involves. Connectionist models have been applied to several developmental disorders, including WS, SLI, autism, developmental dyslexia, and schizophrenia. In these cases, the researcher starts with a model formulated to capture the normal trajectory of development in a given cognitive domain. Over the last fifteen years, connectionist Karmiloff-Smith & Thomas, page 38 models have been applied to many phenomena within normal cognitive development, including those in infancy (e.g., categorisation, object-directed behaviour, memory), in childhood (e.g., Piagetian reasoning tasks such as the balance scale problem, seriation, and conservation), and in language development (e.g., categorisation of speech sounds, segmentation of the speech stream into words, vocabulary development, inflectional morphology, syntax, metaphor, reading). In these models, it is possible to identify the computational constraints that guide the normal trajectory of development, such as the architecture of the connectionist network, the activation dynamics of the system, its input and output representations and its learning rule (Karmiloff-Smith & Thomas, in press; Thomas & Karmiloff-Smith, 2002, in press, a, b, c). Either psychological or neuroscientific data are then used to motivate alterations to these initial constraints in an attempt to capture the atypical trajectory of development observed in a particular disorder, and any endstate deficits in the adult. To the intermediate level of computational modelling, one may import downwards constraints from the psychological level, such as the differences found in phonological and semantic representations of individuals with in WS (Thomas & Karmiloff-Smith, in press-a), or in the phonological representations of individuals with SLI or developmental dyslexia (e.g., Harm & Seidenberg, 1999; Hoeffner & McClelland, 1993). Or one may import upwards constraints from the neural level, such as the differences in neuronal density observed in different areas of the brains of individuals with autism (Cohen, 1998), or differences in the level of dopamine in the frontal lobes of individuals with schizophrenia (Cohen & Servan-Schreiber, 1992). Karmiloff-Smith & Thomas, page 39 One of the advantages of computational models is that they allow candidate explanations of the developmental process to be explicitly formulated, and for the process to be examined under a range of controlled conditions beyond the scope of current empirical methods. For example, in one set of simulations (Thomas & Karmiloff-Smith, in press-b), we compared the effect of the same type of damage applied to a developmental system either prior to training (to represent the case of a developmental disorder) or following training (to represent the case of an adult acquired disorder). This comparison allowed us to assess the contribution of the developmental process (here a learning rule driving the acquisition of a representative cognitive domain) in producing patterns of deficits in the final trained state following various different types of damage. The results revealed a complicated relationship between patterns of deficits following startstate damage and patterns of deficits following endstate damage. For certain types of damage (e.g., lesioning), the system was far more sensitive to damage in the endstate than the startstate – development served to attenuate the effects of differences in the processing structures. For other types of damage (e.g., processing noise), the system was far more sensitive to damage in the startstate than the endstate – development served to exaggerate the effects of differences in the activation dynamics because in the endstate representations were already more stable. In some cases, the effects of damage were global (noise, lesioning), but in other cases the deficit was selective to particular aspects of the cognitive domain only following damage to the endstate. To the extent these networks serve as valid models of development, the results emphasise the importance of the developmental process in determining the patterns of endstate deficits following alteration to different initial neurocomputational constraints. Karmiloff-Smith & Thomas, page 40 In another set of simulations (Thomas & Karmiloff-Smith, in press-b), we examined a developmental system which produced emergent specialisation of particular functions to particular structures during development. This model allowed us to explore the conditions under which static explanations of developmental disorders might hold. That is, if modules are an outcome of development, and one part of the system is damaged prior to training, does the rest of the system nevertheless develop normally? Such a condition (which we term residual normality) must hold if strong analogies are to be drawn between selective cognitive deficits in developmental disorders and those found in cases of adult damage to previously normally developed brains. Our simulations revealed that the conditions under which residual normality would hold are fairly narrow, and in many cases developmentally implausible. A developmental system suffering initial, selective damage tends to use its remaining resources to compensate for the initial damage across development. Our computational work therefore supports the conclusions of Section VI B, where we established that in many cases, focal brain damage in young healthy children is followed by recovery. Computational models can also be applied to particular disorders and specific sets of empirical data. This type of work allows us to evaluate whether particular theoretical claims are actually sufficient to explain the behavioural deficits observed in a given developmental disorder. Thus in separate work (Thomas & Karmiloff-Smith, in pressa), we have explored how a connectionist model of normal development of inflectional morphology (the English past tense) may be applied to the case of WS. This type of a model attempts to maximise the psychological plausibility of its constraints, and fit actual patterns of empirical data, from typically developing children when the constraints are normal and from the target disorder when the Karmiloff-Smith & Thomas, page 41 constraints have been altered from the outset in line with the available empirical evidence. In WS, the evidence on the acquisition of past tense formation initially indicated difficulty with producing irregular inflections (e.g., think-thought, give-gave) (Clahsen & Almazan, 1998). However, these preliminary findings have been hard to replicate, and larger studies have suggested that there exist both problems generalising inflectional regularities to novel word forms (wug-wugged), and a differential influence of lexical semantics on inflection in WS (typically developing children found irregular verbs with more abstract meanings harder to inflect than those with more concrete meanings, but children and adults with WS performed equally on both) (Thomas et al., 2001). In using a computational model to explore this aspect of language acquisition in WS, we pursued the hypotheses discussed in the previous two sections. We explored whether manipulations to the initial phonological and semantic representations within the normal model were sufficient to shift its developmental performance from that of typically developing children to that of our WS cohort. The results showed that insufficiently abstract phonological representations (i.e., with reduced redundancy and between phoneme similarity) were able to capture the problems in inflecting novel forms, and that weakened semantic representations were able to capture poor development of irregular inflection, as well as empirical evidence for differential influence of semantic variables such as abstractness of verb meaning. Importantly, this model demonstrated that to capture the full range of individual variation in the empirical data for individuals with WS, multiple initial neurocomputational Karmiloff-Smith & Thomas, page 42 constraints had to be altered – but that when more than one constraint was altered, the effects on the subsequent developmental trajectory tended to be interactive rather than additive. For example, weaker semantic constraints tended to exaggerate the effects of changes to the phonological constraints. Although models such as these necessarily contain simplifications (the hallmark of any modelling process), we believe that they represent one of the vital ways forward in evaluating developmental deficits with a very concrete notion of the developmental process in mind. Thus, while our model demonstrated deficits at the end of its development that matched patterns found in adults with WS, crucially those deficits were the outcome of differential initial computational constraints and the subsequent process of development, and did not correspond to the simple removal of any preexisting static structures. VII. Conclusion: Contribution to Developmental Theory of the Study of Language in WS - a Natural Experiment of Nature. Consistent functional structure appears to emerge in the cognitive systems of normal adults, and there has long been a debate about the origin of this consistency. At one extreme, there have been theories of pre-wired, innate modular structure, while at the other there have been theories of equipotentiality and structure derived from regularities in the environmental input. Recently, developmental cognitive neuroscience has moved towards a middle way, the idea of emergentism, whereby initial constraints in computational properties in the brain ‘seed’ specialisation that emerges as a product of development (Elman, et al., 1996; Karmiloff-Smith, 1992, 1998; see Thomas, in press, for discussion). However, revealing the nature of these Karmiloff-Smith & Thomas, page 43 constraints is complex, since normal development confounds the consistency of these constraints across the population with the consistency of the environment to which most individuals are exposed. It is thus developmental disorders that may provide a window on these constraints, since they represent a situation where the constraints start out differently, and where cognitive-level deficits may be the eventual outcome of development rather than the initial state. In the case of genetic developmental disorders with uneven cognitive profiles in their outcome, we have started to begin to explore more specific links between genotype and phenotype via case study comparisons (Karmiloff-Smith, Grant, Ewing, Carette, Metcalfe, Donnai, Read & Tassbehji, 2003). Yet, in our view, progress cannot be made if researchers continue to characterise deficits within static models, based on behavioural data from older children and adults (Frangiskakis et al., 1996). This is because the behavioural deficits that arise from genetic mutations must be traced back to their infant origins and to the cognitive level of account, with development as a crucial component of the link (Karmiloff-Smith, 1998; Karmiloff-Smith, Scerif & Ansari, 2003). In this paper, we have illustrated this argument with reference to one disorder, Williams syndrome. We have detailed the genotype, as well as the physical phenotype, and cognitive phenotype. Then, concentrating on the domain of language development, we have shown how static accounts of deficits need to be replaced by developmental accounts. To do so, we have explored similarities to, and differences from, deficits found in cases of child and adult acquired brain damage. A realistic causal model of the link between genes and behaviour led us to examine a range of Karmiloff-Smith & Thomas, page 44 atypical precursors to language development in WS, and then to construct a preliminary developmental account of WS language acquisition. Finally, we stressed the utility of computational modelling for linking cognitive and neural levels in the study of developmental disorders, and for evaluating concrete formulisations of the developmental process. In conclusion, we believe that we have demonstrated that developmental disorders can be highly effective experiments of nature, provided the very process of development itself remains at the heart of the explanation. Karmiloff-Smith & Thomas, page 45 References Anderson, V., Northam, E., Hendy, J., & Wrennall, J. (2001). Developmental Neuropsychology: A Clinical Approach. Hove, Sussex: Psychology Press. Anderson, J. A., Silverstein, J. W., Ritz, S. A., & Jones, R. S. (1977). Distinctive features, categorical perception, and probability learning: Some applications of a neural model. Psychological Review, 84, 413-451. Atkinson, J., Anker S., Braddick, O., Nokes, L., Mason, A. & Braddick, F. (2001). Visual and visuospatial development in young children with Williams syndrome. Developmental Medicine and Child Neurology, 43(5), 330-337. Arber, S., Barbayannis, F.A., Hanser, H., Schneider, C., Stanyon, C.A., Bernard, O., & Caroni, P. (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature, 393, 805-809. Baron-Cohen, S. (1998). Modularity in developmental cognitive neuropsychology: Evidence from autism and Gilles de la Tourette syndrome. In J. A. Burack, R. M. Hodapp, & E. Zigler (Eds.), Handbook of Mental Retardation and Development (pp. 334-348). Cambridge University Press, Cambridge, UK. Bates, E. & Roe, K. (2001). Language development in children with unilateral brain injury. In C.A. Nelson, and M. Luciana (Eds.), Handbook of developmental cognitive neuroscience, (pp. 281-307). Cambridge, MA: MIT. Bearden, C. E., Wang, P. P., & Simon, T. J. (2002). Williams syndrome cognitive profile also characterizes Velocardiofacial/DiGeorge Syndrome. American Journal of Medical Genetics (Neuropsychiatric Genetics), 114, 689-692. Bellugi, U., Wang, P., & Jernigan, T. L. (1994). Williams syndrome: an unusual neuropsychological profile. In S. Broman and J. Grafman (Eds.), Atypical Karmiloff-Smith & Thomas, page 46 cognitive deficits in developmental disorders: Implications for brain function (pp 23-56). Hillsdale, NJ: Erlbaum. Bertrand, J., Mervis, C., Rice, C.E. & Adamson, L. (1993). Development of joint attention by a toddler with Williams syndrome. Paper presented at the Gatlinberg Conference on Research and Theory in Mental Retardation and Developmental Disabilities, Gatlinberg. Beuren, A.J., Apitz, J.& Harmjanz, D. (1962). Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation 26,1235-1240. Bishop, D. V. M. (1997). Cognitive neuropsychology and developmental disorders: Uncomfortable bedfellows. Quarterly Journal of Experimental Psychology, 50A, 899-923 Black, J.A. & Bonham-Carter, R.E. (1963). Association between aortic stenosis and facies of severe infantile hypercalcemia. Lancet, 2, 745-748. Broder, K., Reinhardt, E., Ahern, J., Lifton, R., Tamborlane, W., & Pober, B. (1999). Elevated ambulatory blood pressure in 20 subjects with Williams syndrome. American Journal of Medical Genetics, 83, 356-360. Brown, J., Johnson, M.H., Paterson, S., Gilmore, R.O, Gsödl, M, Longhi, E., & Karmiloff-Smith, A. (2003). Spatial representation and attention in toddlers with Williams syndrome and Down syndrome. Neuropsychologia, 41(8), 1037-1046. Cappelletti, M., Butterworth, B., & Kopelman, M. (2001). Spared numerical abilities in a case of semantic dementia. Neuropsychologia, 39, 1224-1239. Cherniske, E.M., Sadler, L.S., Schwartz, D., Carpenter, T.O., & Pober, B.R. (1999). Early puberty in Williams syndrome. Clinical Dysmorphology, 8, 117-121. Karmiloff-Smith & Thomas, page 47 Cicchetti, D. (2002). The impact of social experience on neurobiological systems: illustration from a constructivist view of child maltreatment, Cognitive Development, 17, 3-4, 1407-1428. Cicchetti, D., & Tucker, D. (1994). Development and self-regulatory structures of the mind. Development and Psychopathology, 6, 533-549. Cipolotti, L., Butterworth, B. & Warrington, E. (1995) Selective impairment of the manipulation of arabic numerals. Cortex, 31, 73-86. Clahsen, H., & Almazan, M. (1998). Syntax and morphology in Williams syndrome. Cognition, 68, 167-198. Clahsen, H. & Almazan, M. (2001). Compounding and inflection in language impairment: Evidence from Williams Syndrome (and SLI). Lingua, 111, 729-757. Cohen, I. L. (1998). Neural network analysis of learning in autism. In D. Stein & J. Ludick (Eds.), Neural networks and psychopathology (pp. 274-315). Cambridge: Cambridge University Press. Cohen, J.D. & Servan-Schreiber, D. (1992). Context, cortex and dopamine: A connectionist approach to behavior and biology in schizophrenia. Psychological Review, 99, 45-77. Cohen, Ansari, Rosen & Karmiloff-Smith (March, 2002). Presentation to the Williams Syndrome Workshop, London. Coltheart, M. (2002). Assumptions and Methods. In B. Rapp (Ed), The Handbook of Cognitive Neuropsychology: What Deficits Reveal About the Human Mind, Psychology Press. Curran, M.E., Atkinson, D.L., Ewart, A.K., Morris, C.A., Leppert, M.F., & Keating, M.T. (1993). The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell, 73,159-168. Karmiloff-Smith & Thomas, page 48 Deruelle, C., Mancini, J., Livet, M.O., Cassé-Perrot, C., de Schonen, S. (1999). Configural and local face processing in children with Williams syndrome. Brain and Cognition, 41, 276-298. Donnai, D. & Karmiloff-Smith, A. (2000) Williams syndrome: From genotype through to the cognitive phenotype. American Journal of Medical Genetics: Seminars in Medical Genetics, 97 (2), 164-171. Einfeld,, S.L., Tonge, B.J., & Florio, T. (1997). Behavioral and emotional disturbance in individuals with Williams syndrome. American Journal on Mental Retardation, 102(1), 45-53. Elman, J. L., Bates, E. A., Johnson, M. H., Karmiloff-Smith, A., Parisi, D., & Plunkett, K. (1996). Rethinking innateness: A connectionist perspective on development. Cambridge, MA.:MIT Press. Ewart, A.K., Morris, C.A, Ensing, G.J., Loker, J., Moore, C., Leppert, M., & Keating, M. (1993a). A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proceedings of the National Academy of Science, 90, 3226-3230. Ewart, A.K., Morris, C.A., Atkinson, D., Weishan, J., Sternes, K., Spallone, P., Stock, D.A., Leppert, M., & Keating, M.T. (1993b). Hemizygosity at the elastin locus in a developmental disorder, Williams Syndrome. Nature Genetics, 5, 11-16. Franke, U. (1999). Williams-Beuren syndrome: genes and mechanisms. Human Molecular Genetics, 8, 1947-1954. Frangiskakis, J.M., Ewart, A.K., Morris, A.C., Mervis, C.B., Bertrand, J., Robinson, B.F., Klein, B.P., Ensing, G.J., Everett, L.A., Green, .D., Proschel, C., Cutowski, N.J., Noble, M., Atkinson, D.L., Odelberg, S.J. & Keating, M.T. (1996). LIMkinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell, 86, 59-69. Karmiloff-Smith & Thomas, page 49 Galaburda, A.M., Wang, P.P., Bellugi, U. & Rossen, M. (1994). Cytoarchitectonic anomalies in a genetically based disorder: Williams syndrome. Cognitive Neuroscience and Neuropsychology, NeuroReport, 5, 753-757. Gosch, A. & Pankau, R. (1997). Personality characteristics and behaviour problems in individuals of different ages with Williams syndrome. Developmental Medicine and Child Neurology, 39(8), 527-533. Grant, J., Karmiloff-Smith, A., Gathercole, S. A., Paterson, S., Howlin, P., Davies, M., & Udwin, O. (1997). Phonological short-term memory and its relationship to language in Williams syndrome. Cognitive Neuropsychiatry, 2, 81-99. Grant, J., Valian, V., & Karmiloff-Smith, A. (2002). A study of relative clauses in Williams syndrome. Journal of Child Language, 29, 403-416. Grice, S., Spratling, M.W., Karmiloff-Smith, A., Halit, H., Csibra, G., de Haan, M., & Johnson, M.H. (2001). Disordered visual processing and oscillatory brain activity in autism and Williams Syndrome. Neuroreport, 12, 2697-2700. Hallidie-Smith, K.A., & Karas, S. (1988). Cardiac anomalies in Williams-Beuren syndrome. Archives of Disease in Childhood, 63, 809-813. Harm, M. & Seidenberg, M. S. (1999). Phonology, reading acquisition, and dyslexia: Insights from connectionist models. Psychological Review, 106, 491-528. Hebb, D. O. (1949). The organization of behavior. New York: Wiley. Hoeffner, J. H. & McClelland, J. L. (1993). Can a perceptual processing deficit explain the impairment of inflectional morphology in developmental dysphasia? A computational investigation. In E.V. Clark (Ed), Proceedings of the 25th Child language research forum. Stanford University Press. Howlin, P., Davies, M. & Udwin, O. (1998a). Cognitive functioning in adults with Williams syndrome. Journal of Child Psychology and Psychiatry, 39, 183-189. Karmiloff-Smith & Thomas, page 50 Howlin, P., Davies, M., & Udwin, O. (1998b). Syndrome specific characteristics in Williams syndrome: To what extent do early behavioural patterns persist into adult life? Journal of Applied Research in Intellectual Disabilities, 11, 207-226. Huttenlocher, P.R. (2002) Neural Plasticity: The effects of environment on the development of the cerebral cortex. Cambridge, MA, US: Harvard University Press. Jarrold, C., Hartley, S. J., Phillips, C., & Baddeley, A. D. (2000). Word fluency in Williams syndrome: Evidence for unusual semantic organisation? Cognitive Neuropsychiatry, 5, 293-319. Jarrold, C., Phillips, C. E., Baddeley, A. D., Grant, J., & Karmiloff-Smith (July, 2001). Comprehension of spatial and non-spatial language in Williams syndrome. Paper presented at the Experimental Psychology Society, Manchester Meeting. Johnson, S. & Carey, S. (1998). Knowledge enrichment and conceptual change in folk biology: Evidence from Williams syndrome. Cognitive Psychology, 37, 156-184. Johnson, M. H., & Morton, J. (1991). Biology and Cognitive Development: The case of face recognition. Oxford: Blackwell. Jones, W, Bellugi, U., Lai, Z., Chiles, M., Reilly, J., Lincoln, A., & Adolphs, R. (2000). Hypersociability in Williams syndrome. Journal of Cognitive Neuroscience, 12: Supplement, 30-46. Karmiloff-Smith, A. (1992). Beyond modularity: A developmental perspective on cognitive science. Cambridge, MA: MIT Press/Bradford Books. Karmiloff-Smith, A. (1997). Crucial differences between developmental cognitive neuroscience and adult neuropsychology. Developmental Neuropsychology, 13, 4, 513-524. Karmiloff-Smith & Thomas, page 51 Karmiloff-Smith, A. (1998). Development itself is the key to understanding developmental disorders. Trends in Cognitive Sciences, 2, 10, 389-398. Karmiloff-Smith, A., Grant, J., Ewing, S., Carette, M.J., Metcalfe, K., Donnai, D., Read, A.P., & Tassbehji, M. (2003). Using case study comparisons to explore genotype/phenotype correlations. Journal of American Medical Genetics, 40: 136140. Karmiloff-Smith, A., Scerif, G., Ansari, D. (2003). Double dissociations in developmental disorders? Theoretically misconceived, empirically dubious. Cortex, 39, 161-163. Karmiloff-Smith, A., Scerif, G., & Thomas, M. S. C. (2002). Different approaches to relating genotype to phenotype in developmental disorders. Developmental Psychobiology, 40, 311-322. Karmiloff-Smith, A. & Thomas, M. (in press). Can developmental disorders be used to bolster claims from Evolutionary Psychology? A neuroconstructivist approach. In J. Langer, S. Taylor Parker & C. Milbrath (Eds.) Biology and Knowledge Revisited: From Neurogenesis to Psychogenesis. Laing, E., Butterworth, G., Ansari, D., Gsödl, M., Longhi, E., Panagiotaki, G., Paterson, S., & Karmiloff-Smith, A. (2002). Atypical development of language and social communication in toddlers with Williams syndrome. Developmental Science, 5, 233-246. Laing, E., Hulme, C., Grant, J., & Karmiloff-Smith, A. (2001). Learning to read in Williams syndrome: Looking beneath the surface of atypical reading development. Journal of Child Psychology and Psychiatry, 42, 729-739. Leslie, A.M. (1992). Pretence, autism, and the Theory-of-Mind-Module. Current Directions in Psychological Science, 1, 18-21. Karmiloff-Smith & Thomas, page 52 Masataka, N. (2000). Information from speech and gesture is integrated when meanings of new words are categorized in normal young children but not in children with Williams syndrome. Bulletin of the Japanese Cognitive Science Society, 7, 37-51. Masataka, N. (2001). Why early linguistic milestones are delayed in children with Williams syndrome: late onset of hand banging as a possible rate-limiting constraint on the emergence of canonical babbling. Developmental Science, 4, 158-164. Mervis, C., & Bertrand, J. (1997). Developmental relations between cognition and language: Evidence from Williams Syndrome. In L. B. Adamson & M. A. Romski (Eds.), Research on communication and language disorders: Contributions to theories of language development (pp. 75-106). New York: Brookes. Mervis, C. B., Morris, C. A., Bertrand, J., & Robinson, B. F. (1999). William Syndrome: Findings from an integrated program of research. In H. Tager-Flusberg (Ed), Neurodevelopmental disorders (pp. 65-110). Cambridge, Mass: MIT Press. Mervis, C.B., Robinson, B.F., Bertrand, J., Morris C.A., Klein-Tasman, B.P. & Armstrong, S.C. (2000). The Williams syndrome cognitive profile. Brain & Cognition, 44, 604-628. Mervis, C.B., Robinson, B.F., Bertrand, J., Morris C.A., Klein-Tasman, B.P., Bonita., P., & Armstrong,, S.C. (2002). The Williams Syndrome Cognitive Profile. Brain and Cognition, 44(3): 604-628 Morris, C.A., Demsey, S.A., Leonard, C.O., Dilts, C., & Blackburn, B.L. (1988). Natural history of Williams syndrome: Physical characteristics. Journal of Paediatrics, 113(3), 18-326. Karmiloff-Smith & Thomas, page 53 Morris, C.A., Thomas, I.T., & Greenberg F. (1993). Williams syndrome: Autosomal dominant inheritance. American Journal of Medical Genetics, 47, 478-481. Nazzi, T., & Karmiloff-Smith, A. (2002). Early categorization abilities in young children with Williams syndrome. NeuroReport, 13(10):1259-1262. Nazzi, T., Paterson, S. & Karmiloff-Smith, A. (2003). Early word segmentation by infants and toddlers with Williams syndrome. Infancy, 4 (2), 251-271 Neville, H. J., Mills, D. L., & Bellugi, U. (1994). Effects of altered auditory sensitivity and age of language acquisition on the development of language– relevant neural systems: preliminary studies of Williams syndrome. In S. Broman and J. Grafman (Eds.), Atypical cognitive deficits in developmental disorders: Implications for brain function (pp. 67-83). Hillsdale, NJ: Erlbaum. Pankau, R., Gosch, A., Simeoni, E., & Wessel, A. (1993). Williams-Beuren syndrome in monozygotic twins with variable expression. American Journal of Medical Genetics, 47, 475-477. Paterson, S. J. (2000). The development of language and number understanding in Williams Syndrome and Down's Syndrome: Evidence from the infant and mature phenotypes. Unpublished doctoral thesis. University College London. Paterson, S. J., Brown, J. H., Gsödl, M. K., Johnson, M. H., & Karmiloff-Smith, A. (1999). Cognitive modularity and genetic disorders. Science, 286, 2355-2358. Patterson, Karalyn E., 1981. Neuropsychological approaches to the study of reading. British Journal of Psychology, 72.151-174. Pennington, B.F. (2001). Genetic methods. In C.A. Nelson and M. Luciana (Eds.), Handbook of developmental cognitive neuroscience (pp.149-158). Cambridge, MA: MIT Press. Karmiloff-Smith & Thomas, page 54 Peoples, R., Franke, Y., Wang, Y.-K., Perez-Jurado, L., Paperna, T., Cisco, M., & Franke, U. (2000). A physical map, including a BAC/PAC clone contig of the Williams-Beuren Syndrome deletion region at 7qll.23. American Journal of Human Genetics, 66, 47-67. Perez-Juralo, L.A., Peoples, R., Kaplan, P., Hamel, B.C.J., & Franke, U. (1996). Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. American Journal of Human Genetics, 59, 781792. Pinker, S. (1991). Rules of language. Science, 253, 530-535. Pinker, S. (1994). The language instinct. London: Penguin. Pinker, S. (1997). How the mind words. New York: Norton. Pinker, S. (1999). Words and rules. London: Weidenfeld & Nicolson. Proschel, C., Blouin, M. J., Gutowski, N. J., Ludwig, R., & Noble, M. (1995). L1MK1 is predominantly expressed in neural tissue and phosphorylates serine, threonine and tyrosine residues in vitro. Oncogene, 11, 1271 – 1281. Rae. C., Karmiloff-Smith, A., Lee, M.A., Dixon, R.M., Blamire, A.M., Thompson, C.H., Grant, J., Styles, P. & Radda, G.K.. (1998) Brain Biochemistry in Williams Syndrome. Evidence for a role of the cerebellum in cognition? Neurology, 51, 3340. Rapp, A., & Caramazza, A. (2002) Selective difficulties with spoken nouns and written verbs: A single case study. Journal of Neurolinguistics, 15, 373-402. Robinson, W.P., Waslynka, J., Bernasconi ,F., Wang, M., Clark, S., Kotzot, D., & Schinzel, A. (1996). Delineation of 7q11.2 deletions associated with WilliamsBeuren syndrome and mapping of repetitive sequences to within and to either side of the common deletion. Genomics, 34,17-23. Karmiloff-Smith & Thomas, page 55 Rossen, M., Klima, E. S., Bellugi, U., Bihrle, A., & Jones, W. (1996). Interaction between language and cognition: Evidence from Williams syndrome. In J. H. Beitchman, N. Cohen, M. Konstantareas & R. Tannock (Eds.), Language Learning and Behaviour (pp. 367-392). New York, NY: Cambridge University Press. Rourke, B. P. (1987). Syndrome of non-verbal learning disabilities: The final common pathway of white matter disease/dysfunction. The Clinical Neuropsychologist, 1, 209-234. Rourke, B. P. (1989). Nonverbal learning disabilities. New York: Guilford Press. Rourke, B. P., & Tsatsanis, K. D. (1996). Syndrome of nonverbal learning disabilities: Psycholinguistic assets and deficits. Topics in Language Disorders, 16, 30-44. Scott, P., Mervis, C. B., Bertrand, J., Klein, B. P., Armstrong, S. C., & Ford, A. L. (1995). Semantic organization and word fluency in 9- and 10-year-old children with Williams syndrome [special issue]. Genetic Counseling, 6, 172-173. Shallice, T. (1988). From neuropsychology to mental structure. Cambridge: Cambridge University Press. Singer Harris, N. G., Bellugi, U., Bates, E., Jones, W., & Rossen, M. (1997). Contrasting profiles of language development in children with Williams and Down syndromes. Developmental Neuropsychology, 13, 345-370. Smith, N. (1999). Chomsky: Ideas and ideals. Cambridge: Cambridge University Press. Smith, N. & Tsimpli, I-M. (1995). The Mind of a Savant: Language, Learning and Modularity, Oxford: Blackwell. Karmiloff-Smith & Thomas, page 56 Stevens, T., & Karmiloff-Smith, A. (1997). Word learning in a special population: do individuals with Williams syndrome obey lexical constraints? Journal of Child Language, 24, 737-765. Tager-Flusberg, H., Boshart, J., & Baron-Cohen, S. (1998). Reading the windows to the soul: Evidence of domain-specific sparing in Williams syndrome. Journal of Cognitive Neuroscience, 10(5), 631-639. Tassabehji, M., Metcalfe, K., Fergusson, W.D., Carette, M.J., Dore, J.K., Donnai, D., Read, A.P., Proschel, C., Gutowski, N.J., Mao, X., & Sheer, D. (1996). LIM-kinase deleted in Williams syndrome. Nature Genetics, 13, 272-273. Temple, C. (1997). Developmental cognitive neuropsychology. Psychology Press. Temple, C., Almazan, M., & Sherwood, S. (2002). Lexical skills in Williams syndrome: a cognitive neuropsychological analysis. Journal of Neurolinguistics, 15(6), 463-495. Temple, C & Clahsen, H (in press). How connectionist simulations fail to account for developmental disorders in children. Behavioral and Brain Sciences. Thomas, M. S. C. (in press). Multiple causality in developmental disorders: Methodological implications from computational modelling. Developmental Science. Thomas, M. S. C., Dockrell, J., Messer, D., Parmigiani, C., Ansari, D., & KarmiloffSmith, A. (2002). Naming in Williams syndrome. Manuscript submitted for publication. Thomas, M. S. C., Grant, J., Gsödl, M., Laing, E., Barham, Z., Lakusta, L., Tyler, L. K., Grice, S., Paterson, S. & Karmiloff-Smith, A. (2001). Past tense formation in Williams syndrome. Language and Cognitive Processes, 16, 143-176. Karmiloff-Smith & Thomas, page 57 Thomas, M. S. C. & Karmiloff-Smith, A. (2002). Modelling typical and atypical cognitive development. In U. Goswami (Ed.), Handbook of Childhood Cognitive Development (pp. 575-599). Oxford: Blackwell Publishers. Thomas, M. S. C. & Karmiloff-Smith, A. (in press, a). Modelling language acquisition in atypical phenotypes. Psychological Review. Thomas, M. S. C. & Karmiloff-Smith, A. (in press, b). Are developmental disorders like cases of adult brain damage? Implications from connectionist modelling. Behavioral and Brain Sciences. Thomas, M. S. C. & Karmiloff-Smith, A. (2003). Connectionist models of development, developmental disorders and individual differences. In R. J. Sternberg, J. Lautrey, & T. Lubart (Eds.), Models of Intelligence: International Perspectives, (p. 133-150). American Psychological Association. Thomas, M. S. C., van Duuren, M., Ansari, D., Parmigiani, C., & Karmiloff-Smith, A. (2002). The development of semantic categories and metaphor comprehension in Williams syndrome. Manuscript in preparation. Udwin, O. & Yule, W. (1991). A cognitive and behavioural phenotype in Williams syndrome. Journal of Clinical and Experimental Neuropsychology, 13, 232-244. Urban. Z., Helms, C., Fekete, G., Csiszar, K., Bonnet, D., Munnich, A., Donis-Keller, H., & Boyd, C.D. (1996). 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. American Journal of Human Genetics, 59, 958-962. Volterra, V., Capirci, O., & Caselli, M. C. (2001). What atypical populations can reveal about language development: The contrast between deafness and Williams syndrome. Language and Cognitive Processes, 16, 219-239. Karmiloff-Smith & Thomas, page 58 Wexler K (1996). The development of inflection in a biologically based Theory of language acquisition. In Toward a Genetics of Language, ML Rice (Ed). Mahwah, NJ: Lawrence Erlbaum Associates, Inc. Williams, J.C.P., Barratt-Boyes, B.G. & Lowe, J.B. (1961). Supravalvular aortic stenosis. Circulation, 24, 1311-1318. Winter, M., Pankau, R., Amm, M., Gosch, A,, & Wessel, A. (1996). The spectrum of ocular features in the Williams-Beuren syndrome. Clinical Genetics, 49, 28-31. Zukowski, A. (2001). Uncovering grammatical competence in children with Williams syndrome. Unpublished doctoral thesis, Boston University. 1 We thank our colleague, Julia Grant, for drawing our attention to the fact that our original + sign implied an additive notion whereas we had in mind a multiplicative notion. In fact, more precisely the equation should read f(x, y, z) where x is the initial existing neurocomputational state, y defines the way it changes in response to activity, and z is the internal/external environment. But the equation used above is more self explanatory. 2 Whether the subsystem falls inside or outside the normal range of function is a dichotomy enforced by measurement on a standardised test. Further, more subtle measures are required to establish the degree to which functional structure actually falls within the normal range of variation.