NMRDocumentation
advertisement

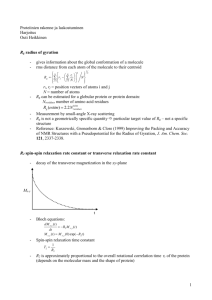
NMR Documentation This document describes how the programs are to be used. The inputs and outputs are described. The example output is given in the directory Example. These programs should be of use to anybody who wants to use trajectories. Programs Inputs and outputs Example command lines for each of the programs Bash files with example commands Example output Interpretation of the output Documentation explaining the theory of the calculations of the programs The 3J programs are - 3J_coupling_calculation - 3J_coupling_check The noe programs are - noe_calculation - noe_check There is also a program that calculates the text files to calculate the noe at any mixing time - noecalculation There is a program to examine the trajectory - image_of_trajectory The relaxation rate programs are - relaxation_rate - particular_relaxation_rate - particular_relaxation_rate_no_tumbling There are also bash files for these programs. 3J_coupling_calculation This program calculates the average 3J couplings of a trajectory. The 3J coupling of each frame is calculated of the atom-atom-atom-atom, than it averages over all the frames. The default output is the C-…-…-C, C-…-…-H, H-…-…-C, H-…-…-H 3-bonds. The atoms are adjacent. - Types of coefficients and functions The couplings are found as a function of the coupling function, e.g. Acos*cos+Bcos+C. The program uses a file called fileequation.txt which has the information of the 3J couplings. This file is parsed for each of the calculations to determine the 3J coupling coefficients. The user can add to the file any coefficients for any type of 3J coupling. The file has all the coefficients of the 3J couplings for each of the 4 adjacent atoms. An example of coefficients for a ‘type’ of 3J coupling is in fileequation.txt Karplus C C C C 3.49 0 .16 The program will open the fileequation.txt file, look to find if the user type input (e.g. Karplus) is found, than parse the information for the 1-4 atoms and coefficients. In this example, if the user wanted a Karplus type of 3J coupling for the C-C-C-C then the coefficients of A=3.49, B=0, C=.16 are used in the 3J coupling calculation. - Theory of the 3J coupling calculation The paper “Developments in the Karplus equation as they relate to the NMR coupling constants of carbohydrates,” Advances in Carbohydrate Chemistry and Biochemistry, v.62, pp.17-82 describes the theory of the 3J couplings. The fileequation.txt as a default has the coefficients of the Karplus type from that paper. The paper is included in the download. Figure 1 shows the 3J coupling as a function of angle for the Karplus type. There are two angles which give the same 3J coupling. This makes it difficult to use the 3J coupling for determination of structure unless noe’s are also used. - Command line The most basic usage of the program uses ./3J_coupling_calculation file.top file.crd Type out-dir samplefrequency (1) There are also the options of using the command line of ./3J_coupling_calculation file.top file.crd Type out-dir samplefrequency atomType1 atomType2 atomType3 atomType4 ./3J_coupling_calculation file.top file.crd out-dir samplefrequency atomType1 atomType2 atomType3 atomType4 coeff1 coeff2 coeff3 - Input The inputs are described. Topology file - file.top – topology file. This is an amber .prmtop file. Trajectory file - file.crd – trajectory file. This is a trajectory file .crd. Type of 3J calculation - Type – type of coupling – string, e.g. Karplus. This is the type of Karplus coupling as found in fileequation.txt. Output directory of the files - out-dir – the directory of the output, e.g. test. This is the directory in which the output will be given. Sampling – 1 frame out of samplefrequency frames is used. This can be important to uncorrelate the motion. - samplefrequency In case the second optional command line is used the atom types are given as inputs. The third optional command line does not specify a type of Karplus coefficients. The user specifies coeff1, coeff2, coeff3 as inputs. This option could be useful if somebody would like to test different coefficients. - Output The use of the command line (1) will produce files fileCC.txt, fileCH.txt, fileHH.txt. The 1-4 adjacent atoms with type [C=atom1, any=atom4; any=atom1, C=atom4], [C=atom1, any=atom2, any=atom3, Hatom4], [H=atom1, any=atom2, any=atom3, C=atom4], [H=atom1, any=atom2, any=atom3, H=atom4] are used to find all the 3J couplings of the molecule. Example output of the proton-proton 3J coupling is 6 5 7 8 : H-C-C-H 3YA:H1 3YA:C1 3YA:C2 3YA:H2 3.44614 The first four numbers are the atom numbers. The atomTypes are given. The second line says what the residues and atom names of the four atoms. The nomenclature uses the amber format from the topology file. The last number is the average 3J coupling using the coefficients that the user wanted from the fileequation.txt. 3J_coupling_check In order to compare theory with experiment it is necessary to know if the calculations are correct. This program will examine the correlation of 3J couplings as a function of the time of the trajectory. The program will output information that can be used to tell you about the 3J couplings of the 1-4 atoms as a function of time. It calculates the ‘std-mean’ of the 3J couplings. It also calculates the average of the 3J coupling as a function of the trajectory time. These calculations are important to determine if the trajectory is not behaving the way it should. An example would be if the trajectory was made with unnatural constraints. This would show in these calculations in the statistics of the 3J coupling as a function of the time of trajectory. These calculations can also show processes that are present in the motion of the molecule. These could be important. - Theory of the standard deviation in the mean Standard deviation about an average calculation does not make sense if the information does not satisfy the central limit theorem. Statisticians use different techniques to examine sets of information which is correlated. The program uses a binning of the information of the 3J coupling as a function of the trajectory time to calculate the ‘standard deviation in the mean,’ which is a good way to find out how precise the calculated 3J coupling is. Two powerpoint presentations are given which describe the calculations that the program does. The figures that the program give should ‘flatten’ for large number of bins. If the figures don’t flatten, then the trajectory should be examined. Example output of the program is given in figure 1 and figure 2 which show - Bad trajectory - Unusual phenomena such as several processes of different timescales affecting the 3J average calculation The program uses the 3J couplings as a function of the time units than bins them into some number of bins which is specified by the user (bininitial to binfinal). The standard deviation/sqrt(number of bins) of the 3J couplings of the bins is found, as a function of the number of bins. Then a curve fit is done of the standard deviation/sqrt(number of bins) as a function of the number of bins (e.g., a+bx, x=number of bins). First a line is used. Then it uses a quadratic, then a cubic, ..., until the chi square is accurate. - Command line ./adjacentfit file.top file.crd type samplefrequency atomResidue1 atom Name1 atomResidue2 atomName2 atomResidue3 atomResidue4 bininitial binfinal point font - Input Topology file - file.top – topology file Trajectory file - file.crd – trajectory file Type of 3J coupling, e.g. Karplus (2) - type – type of 3J coupling coefficients, e.g. Karplus Samples 1 out of samplefrequency frames - samplefrequency AtomResidues, AtomNames - atomResidue1 – first atom residue name, uses the topology file - atomName1 – first atom name - same for all the atomResidue, atomName Bin information - bininitial, binfinal – the calculation will use the # of bins from bininitial to binfinal, e.g. 2, 100 Figures show this many points - point – the figures will show point of the calculation. The figures should not show 100,000 points of information, typical is 500 for a figure. The program calculates the 3J coupling using the samplingfrequency. The figures only show point data points. E.g., 100,000 frames, samplefrequency=10, point=500. 100,000/10=10,000 frames. The figure of the 3J coupling will show the 3J coupling at every 200 frames. Output type of figures - font – jpeg, eps, epslatex, pdf to be used in the figures - Output The program will produce a figure of the 3J coupling as a function of the trajectory time for the four atoms, a figure of the std-mean as a function of bins, the file of the binning calculation, text file of the 3J coupling as a function of the trajectory time, and a text file of the std-mean as a function of bins. The information of these files can be used to interpret the 3J calculation. noe_calculation This program calculates the noe’s. This calculation is the most time-consuming of the programs. The program will give the noe intensities for all the mixing times specified by the user. These intensities have to be normalized to compare with experiment. The output of this program is what the experimentalists measure. - Theory of calculation The calculation of a noe between two atoms involves all the atoms in the molecule if the complete relaxation rate formalism is used. That is the reason the calculation takes time. The complete relaxation rate matrix can be large. This is an abstract of a presentation which explains the use of the program. NOEs from cross-relaxation between proton pairs provide essential structural information on both large and small molecules. For rigid molecules, the analysis can be straightforward using the inverse sixth power and isolated spin-pair approximation. However, for flexible molecules that have multiple conformational states, such as oligosaccharides, small peptides or intrinsically disordered proteins, simple 1/r^6 averaging over conformational states may not provide structurally relevant models. Although formal treatments of this situation have been known for some time, it is difficult to apply these experimentally. One way is to make use of advances in computational power to generate long molecular dynamics trajectories. These calculated molecular motions can be used to generate correlation functions for dipolar interactions and then treat the system with a full relaxation matrix approach. In general the correlation function has two 1/r^3 terms that are typically combined into 1/r^6 averaging, assuming motions are not correlated. It is not uncommon for small molecules to have internal motions such that the effective inter-nuclear distance is between the centers of motional range of the two nuclei. However, the steep 1/r^6 averaging will heavily weight the positions of closest approach. Time points in an MD trajectory can be used to calculate the correlation function directly, if the trajectory is long enough to sample a uniform distribution of molecular orientations and conformations that would average the NMR data. For small molecules such as a disaccharide, these simulations would be on the order of hundreds of nanoseconds or millisecond range. The paper “Influence of Rapid Intramolecular Motion on NMR Cross Relaxation Rates: A Molecular Dynamics Study of Antamide in Solution,” R. Bruschweller, B. Roux, M. Blackledge, C. Greisenger, M. Karplus, R.R. Ernst, J. Am. Chem. Soc. 1992, 114, 2289-2302 is used for the formulation of the calculation. - Comparing the output of the program with experimental information The model which is used should produce noe intensities which satisfy 𝑛𝑜𝑒1 < 1⁄𝑟16 > = 𝑛𝑜𝑒2 < 1⁄ 6 > 𝑟2 (3) Noe1 and noe2 are the noe intensities of two spin pairs. The expectation values are the average of < 1⁄ > of the trajectory. The noe1 and noe2 intensities are calculated. These are measurable. The 𝑟6 program also calculates the averages of < 𝑟 > and < 1⁄ 6 >, which is included in file_distance.txt. 𝑟 The second noe is used to normalize to compare theory with experiment. This noe is usually found from a spin pair inside a ring, or monomer. The first noe could be from a spin pair across the glycosidic linkage. The second noe has motion which does not change in conformations, i.e. this spin par is inside a ring. The output of the program can be used to compare theory with experiment by normalizing the noe of interest with the second spin pair. An example is given from a GAG. The H3-H5 spin pair in a ring is used to normalize the H1-H3 glycosydic spin pair H1-H3. The simulations are bad by a factor of 3.0 which is about 10% in the interproton distance if the equation (3) is used. Experiment noe Simulation H1-H3 H3-H5 3.6057 8.9491 .4029 H1-H3 H3-H5 .06487 .04828 1.3436 intensity This molecule has two conformations of the rings containing the H1 and the H3. The molecular dynamics simulation in general is known not to be accurate in the population of the conformations. The H1-H3 noe could change to be experimentally correct if the population of these two conformations changed by a factor of 2. Factor of 2-3 is about 10% in the inter-proton distance of the H1-H3 if the sixth root is taken. - noe calculation Figure 3 shows the steps necessary to calculate the noe’s. The topology file is used to simulate the molecule. The correlation function of the spin pair is calculated. This gives information of the tumbling. The fourier transform of the correlation function is calculated. The fourier transform is used to calculate the relaxation rates at some strength of magnetic field. The rho and sigma’s are then used in the complete relaxation matrix, which is exponentiated to find the noe at any mixing time. Figure 3 . - Command line ./noe_calculation file.top file.crd totaltime displaytime samplefrequency bfieldz fourier total_time unit distanceatom font all-notall dir-out - Input Time of simulation, - double totaltime. This is in nanoseconds. Correlation function is displayed for this time - double displaytime. Sampling – 1 frame out of samplefrequency is used to calculate the correlation function - int samplefrequency Magnetic field in Tesla - double bfieldz Fourier transform for display – don’t show all points (3) - double fourier Mixing time is from 0 to total_time ms. - double total_time The figures of the build-up curves show the information in units of unit ms - double unit The spin pairs which are separated by at most distanceatom are included in the complete relaxation matrix. Most noe’s are not measurable if the inter-dstance is less than 5 angstroms. - double distanceatom Output is either all the noes within some distance or particular spin pairs. The output could be >1000 files if the molecule has >100 atoms. all means all the information of the spin pairs are the output. If notall is used, than a file called atom_file.txt is used to output the noe information of those spin pairs. - argv[11]=notall,all The output - dir-out = argv[12] Font of the figures, jpeg, eps, epslatex, pdf - font=argv[13] - Output The output of the program will give many files. Each of the noe’s which are calculated will give a figure of the build-up curve and a txt file of the build-up curve points. Several files are also given which contain the information of the cross-relaxation-matrix, the distances of the spin pairs, and the tumbling times of the spin pairs. These are called - cross_relaxation.txt - file_distance.txt - file_tumbling.txt Cross_relaxation.text has all the information of the rhos and sigmas. File_distance.txt has the distance information of < 𝑟 >, < 1⁄ 6 >, 𝑟 −1⁄6 √1⁄𝑟 6 . File_tumbling has the tumbling in nanoseconds of the spin pairs. Curve fitting of the correlation function is used to determine the tumbling time. An exponential fit is used. Figure 4 shows a build-up curve which is calculated from the complete relaxation matrix. The text file noecurve_OME_H3-OME_H1.txt contains all the data points. The build-up curve is evaluated at some mixing time, e.g. 400 milliseconds, to compare theory with experiment. Figure 4 noecheck This program checks the noe calculation. The program will calculate the average vector distance of a spin pair, which should be 0 if the molecule is tumbling isotropically. It calculates the inter-distance of a spin pair which is used for conformation identification. The program also calculates the std-mean of these quantities to determine if any correlation or unusual phenomena is present of the spin pair. There are several reasons to check the noe calculation, bad trajectory unusual phenomena in the dynamics of the molecule the noe is sensitive to the population of conformations If the trajectory was not made correct then the correlation function used in the calculation of the noe may be bad behaved. A non-isotropic box with the wrong box size or an isotropic box with the wrong size could make the correlation function not compatible with the model used to calculate the noe’s. The dynamics of the molecule may be interesting. The statistics of the 3J coupling shows sometimes that there are processes of different time-scales. These processes could affect the noe calculation. The conformations will add differently to the spin pair noe calculation. The conformations of the spin pair of minimal distance will add most to the noe calculation. These calculations can tell you how precise the trajectory noe calculation is. The measurements are usually 10%. The program will also show how to change the population of the conformations to agree with experiment. - Command line ./noe_check file.top file.crd totaltime samplefrequency residue1 atom1 residue2 atom2 point out-dir font - Input Time of the simulation, nanoseconds. - double totaltime Sample frequency – 1 out of samplefrequency frames used to calculate - int amplefrequency Atom names, residues. - string residue - string atom Point shows this many points in the figures int point Out-dir string arv[10] test –figures are jpeg, eps, epslatex, pdf string font; - Output The program gives information of the distances, the average distances, and the std-mean of the calculation of the distances for the spin-pair. - Distances: The program calculates the distance between the spin pairs. The vector of the spin pair used. The point of the second atom minus the point of the first atom is the vector. There is an x-component of the vector, a y-component, and a z-component. Also there is the magnitude of the vector distance. The output is a figure of these 4 quantities as a function of the trajectory time. The text file with the data points are given in case the figure is not in the form which is wanted; you can replot using this information. The information can be used to determine if the molecule is tumbling isotropically. The averages of these components are given in the figures. Isotropic tumbling should give an average of zero in the vector components. It can be the case if the simulation was done wrong that the average won’t be zero, than the figures and the .txt files will show what is wrong. Figure 5 shows the x-direction for a gag spin pair, UYA:H3 to OME:H1. Average distances The average vector distance of the spin pairs should average to zero if the molecule is tumbling isotropically. The program calculates the average vector distance. This is calculated by: at each time of a trajectory it uses all previous time units to calculate the average vector distance. The average distances are shown in figures for each of the components. The program also calculates the average of the magnitude of the vector distance as a function time. Figures are given also the text file with all the Figure 7 data points of the figure. Figure 7 is an example. The average goes to zero. - Std-mean correlation The correlation of the motion is calculated using the std-mean. This is done for the x-component, ycomponent, z-component, and magnitude of the distance. The output is the figure of the binning, the text file of the data points, and the binning information. This is done for all vector components and the magnitude of the vector of the spin pair. image_of_trajectory This program calculations the inter-distance of a spin pair and shows it in a figure. The program will do this for all trajectories in the directory. - command line .image_of_trajectory topolog trajectory.crd in-dir totaltime out-dir points - Inputs Topology is the topology file of the trajectory. - Toology Trajectory is the trajectory . - Trajectory In-dir i- of the topology/trajectory. Totaltime is the time of the simulation. - Totaltime Out-dir – of the calculation. Points is the number of points shown in the trajectory of the totaltime. - Output The program calculates the inter-distances of the spin pairs. It will give images and text. relaxation_rate This program takes in the topology file and the trajectory file to calculate relaxation rates. Command line ./relaxation_rate file.top file.crd totaltime displaytime samplefrequency bfieldz fourier distanceatom font all-notall dir-out - Input Time of simulation, - double totaltime. This is in nanoseconds. Correlation function is displayed for this time - double displaytime. Sampling – 1 frame out of samplefrequency is used to calculate the correlation function - int samplefrequency Magnetic field in Tesla - double bfieldz Fourier transform for display – don’t show all points - double fourier The spin pairs which are separated by at most distanceatom are included in the complete relaxation matrix. Most noe’s are not measurable if the inter-dstance is less than 5 angstroms. - double distanceatom Output is either all/not-all the relaxation rates within some distance or particular spin pairs. aAll means all the information of the spin pairs are the output. If notall is used, than a file called atom_file.txt is used to output information of those spin pairs. - argv[11]=notall,all The output - dir-out = argv[12] Font of the figures, jpeg, eps, epslatex, pdf - font=argv[13] Output – The file relaxation_rate.txt contains all the proton and carbon relaxation rates. particular_relaxation_rate This program takes in the topology file and the trajectory file to calculate relaxation rates of a spin pair. Command line ./particular_relaxation_rate file.top file.crd totaltime displaytime samplefrequency bfieldz fourier distanceatom font dir-out residue1 atom1 residue2 atom2 - Input Time of simulation, - double totaltime. This is in nanoseconds. Correlation function is displayed for this time - double displaytime. Sampling – 1 frame out of samplefrequency is used to calculate the correlation function - int samplefrequency Magnetic field in Tesla - double bfieldz Fourier transform for display – don’t show all points - double fourier The spin pairs which are separated by at most distanceatom are included in the complete relaxation matrix. Most noe’s are not measurable if the inter-dstance is less than 5 angstroms. - double distanceatom Output is either all/not-all the relaxation rates within some distance or particular spin pairs. aAll means all the information of the spin pairs are the output. If notall is used, than a file called atom_file.txt is used to output information of those spin pairs. - argv[11]=notall,all Font of the figures, jpeg, eps, epslatex, pdf - font=argv[2] The output - argv[13]=out-dir Output – The file carbon_relaxation_rate.txt and proton_relaxation_rate.txt contains all the proton and carbon relaxation rates. There are also figures of the correlation function and fourier transform. There are also text files. particular_relaxation_rate_no_tumbling This program takes in the topology file and the trajectory file to calculate relaxation rates of a spin pair of a correlatin function of the internal motion. The program will average the correlation function of all directions of the B-field. Command line ./particular_relaxation_rate file.top file.crd totaltime displaytime samplefrequency bfieldz fourier distanceatom font dir-out residue1 atom1 residue2 atom2 - Input Time of simulation, - double totaltime. This is in nanoseconds. Correlation function is displayed for this time - double displaytime. Sampling – 1 frame out of samplefrequency is used to calculate the correlation function - int samplefrequency Magnetic field in Tesla - double bfieldz Fourier transform for display – don’t show all points - double fourier The spin pairs which are separated by at most distanceatom are included in the complete relaxation matrix. Most noe’s are not measurable if the inter-dstance is less than 5 angstroms. - double distanceatom Output is either all/not-all the relaxation rates within some distance or particular spin pairs. aAll means all the information of the spin pairs are the output. If notall is used, than a file called atom_file.txt is used to output information of those spin pairs. - argv[11]=notall,all Font of the figures, jpeg, eps, epslatex, pdf - font=argv[2] The output - argv[13]=out-dir Output – The file carbon_relaxation_rate.txt and proton_relaxation_rate.txt contains all the proton and carbon relaxation rates. There are also figures of the correlation function and fourier transform. There are also text files.