Third week
advertisement

Drugs Used in Treatment of Malaria
- Malaria is an infectious disease of humans caused by eukaryotic
protozoon of the genus Plasmodium.
- It is widespread in tropical and subtropical regions, including parts of
the Americas, Asia, and most of Africa.
- The disease results from the multiplication of malaria parasites within
RBCs.
- Malaria remains the world's most devastating human parasitic
infection. Malaria affects over 40% of the world's population. WHO,
estimates that there are 300 - 500 million cases of malaria
worldwide,
- 1-3 million died annually, ~ 90% of them occur among children under
five years of age.
- Four species of Plasmodium can infect and be transmitted by
humans. Severe disease is largely caused by P. falciparum which is
responsible for about 80-90% of the deaths from malaria.
- Malaria caused by P. vivax, P. ovale and P. malariae is generally a
milder disease that is rarely fatal.
- A newly discovered fifth species, P. knowlesi, causes malaria in
monkeys but can also infect humans.
- P. falciparum accounts for 90% of deaths due to malaria and vivax is
the most widely spread species because it exists in both temperate
and tropical climates.
- The malaria life cycle is a complex system with both sexual and
asexual aspects .
Pathogenesis:
- The disease is transmitted to human via biting from the insect vector
is the female Anopheles spp. mosquitos, which breeds in stagnant
water, and the disease it spreads is one of the major killers on our
planet.
- In plasmodium life cycle, man represents the (secondary) hosts,
while female mosquito represents the definitive (primary) host. Only
female mosquitoes feed on blood while male mosquitoes feed on
plant nectar, thus males do not transmit the disease.
- Plasmodium has a complex life cycle that part of it happened within
the mosquito body and the rest of it happened within the human
body
- The life cycle of all species that infect humans is basically the same.
- The part of the life cycle in the mosquito is called “sporogenic cycle”.
It is exogenous phase during which the parasite multiplies to give
sporozoites (infective stage) and it includes both sexual and asexual
formss of multiplication .
- There is also an endogenous asexual phase that takes place in the
vertebrate or human host that is called “schizogenic cycle”.
- This phase includes the parasite development that takes place in the
RBCs, called the “erythrocytic schizogeny” and the phase that takes
place in the parencymal cells in the liver, called the “pre-erythrocytic
schizogeny”.
- The exo-erthrocytic phase is also called the tissue cycle.
1- Human pre-erythrocytic schizogony
- Period of Pre-erythrocytic cycle: on average it take 6-16 days post
infection for different types of plasmodium to complete the preerythrocytic cycle (P. vivax: 8 days, P. falciparum : 6 days, P. malariae
: 13 – 16 days and P. ovale 9 days).
- In certain forms of malaria (P. vivax, P. ovale) some sporozoites
entering the liver cells form hypnozoites, or dormant 'sleeping' forms
of the parasite, which can be reactivated months or years later to
continue an exo-erythrocytic cycle of multiplication.
-
-
-
2- Human erythrocytic schizogony
The librated merozoits invade the RBCs. Following mitotic replication
of its nucleus, the merozoit in the RBCs is termed a schizont. Another
phase of multiplication results in the production of further
merozoites, which are released when the red cell ruptures.
These merozoites then bind to and enter fresh red cells, and the
erythrocytic cycle starts all over again.
During maturation within the red cell, the parasite remodels the host
cell, inserting parasite proteins and phospholipids into the red cell
membrane. The host's haemoglobin is digested and transported to
the parasite's food vacuole, where it provides a source of amino
acids. Free haem, which would be toxic to the plasmodium, is
rendered harmless by polymerisation to haemozoin.
Some antimalarial drugs act by inhibiting the haem polymerase
enzyme responsible for this step.
Typical symptoms of malaria include fever, shivering, arthralgia (joint
pain), vomiting, anemia (caused by hemolysis), hemoglobinuria, and
convulsions.
- The classic symptom of malaria is cyclical occurrence of sudden
coldnessand shivering followed by rigor and then fever (may exceed
40C) and sweating lasting 4-8 hours.
- These stages of malaria symptoms recur at regular intervals
corresponding with synchronous release of merozoites from RBC (48-
72 hrs). Most of malarial parasites causing tertian malaria ('every
third day') .
- Some liberated merozoites from RBCs undergo another
developmental process to form motile intracellular parasites termed
trophozoites.
- This tophozoites differentiate into Male and female gametocytes
(gametogony).
- Macrogametocytes also called female gametocytes
microgametocyte also called as male gametocytes
while
- Some liberated merozoites from RBCs undergo another
developmental process to form motile intracellular parasites termed
trophozoites.
- This tophozoites differentiate into Male and female gametocytes
(gametogony).
- Macrogametocytes also called female gametocytes
microgametocyte also called as male gametocytes
while
3- Mosquito sporogony (Sexual cycle)
- The mosquito becomes infected when it takes a blood meal from an
infected human.
- Once ingested, the parasite gametocytes will further differentiate
into male or female gametes. and then fuse (sexual reproduction) in
the mosquito's gut. This produces an a zygote (ookinete) that
penetrates the gut lining and produces an oocyst in the gut wall.
- When the oocyst ruptures, it releases sporozoites that migrate
through the mosquito's body to the salivary glands, where they are
then ready to infect a new human host.
- The periodic episodes of fever that characterise malaria result from
the synchronised rupture of red cells with release of merozoites and
cell debris.
- The rise in temperature is associated with a rise in the plasma
concentration of TNF-α.
- Recurence of malaria are likely to occur after treatment for three
reasons.
- Recrudescence occurs when parasites are not cleared by treatment,
- Reinfection with new infection established from a separate infective
mosquito bite; both can occur with any malaria parasite species.
- Relapse is specific to P. vivax and P. ovale and involves re-emergence
of blood-stage parasites from latent parasites (hypnozoites) in the
liver. (exoerythrocytic cycle) after an interval of weeks or months to
start the infection again.
Treatment of malaria
The best way to eliminate malaria is to avoid the disease in the first place by
eradicate the vector (Anopheles mosquito) in addition to use the
antimalarial drugs.
Several classes of antimalarial drugs are available . antimalarial drugs are
classified in terms of the action against the different stages of the life cycle
of the parasite.
- Drugs used to treat the acute attack of malaria act on the
parasites in the blood (blood schizonticides) act on the
parasites in the blood (blood schizonticides) e.g. Quinine ,
Mefloquine, Chloroquine, Halofantrine, Sulfones, Pyrimethamine,
and Artemether. Combinations of these agents are frequently used.
Some antibiotics, such as Tetracycline and Doxycycline have proved
useful when combined with the above agents. They act on the
erythrocytic forms of the plasmodium. In infections with P.
falciparum or P. malariae, which have no exoerythrocytic stage, these
drugs effect a cure; with P. vivax or P. ovale, the drugs suppress the
actual attack, but exoerythrocytic forms can re-emerge later to cause
relapses.
- Drugs used for radical cure are active against parasites in
the liver (tissue schizonticides). These agents’ effect a 'radical'
(in the sense of striking at the root of the infection) cure by acting on
the parasites in the liver e.g. Primaquine and Tafenoquine.
- Drugs act on gametocytes and prevent transmission by the
mosquito (gametocides) and thus preventing the increase of the
human reservoir of the disease e.g. Primaquine, Proguanil and
Pyrimethamine .
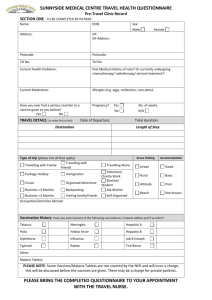
- Drugs used for chemoprophylaxis act on merozoites emerging
from liver cells at the end of the pre-erythrocytic stage (causal
prophylactic drugs). They also block the link between the
exoerythrocytic stage and the erythrocytic stage, and thus prevent
the development of malarial attacks. Chemoprophylactic agents are
mostly given to individuals who intend travelling to an area where
malaria is endemic. Administration should start 1 week before
entering the area and should be continued throughout the stay and
for at least a month afterwards. e.g. Chloroquine, Mefloquine,
Proguanil, Dapsone , Primaquine and Doxycycline. They are often
used in combinations.
-
Major Anti-malarial Drugs
(1) 4-aminoquinoline drivatiives
•
•
•
•
Chloroquine is a synthetic 4-aminoquinoline dr. formulated mainly as
the phosphate salt for oral use.
It is effective against the erythrocytic forms (blood schizonticides) of all
four plasmodial species (if sensitive to the drug), but it does not have
any effect on sporozoites, hypnozoites or gametocytes.
It is considered a drug of choice in treatment of acute attack even
resistance against chloroquine is a growing problem in most parts of
the world especially of P. falciparum and P. vivax .
. Chloroquine is also used as antirheumatoid drug.
Mechanism of action
It is uncharged at neutral pH of the blood and can therefore diffuse freely
into the parasite lysosome. At the acid pH of the lysosome, it is converted
to a protonated, membrane- impermeable form and is 'trapped' inside the
parasite. chloroquine acts mainly on haem disposal by preventing digestion
of haemoglobin by the parasite and thus reducing the supply of amino
acids necessary for parasite viability. It also inhibits haem polymerase that
polymerises toxic free haem to haemozoin rendering it harmless to the
parasite.
Resistance appears to result from enhanced efflux of the drug from parasitic
vesicles as a result of mutations in plasmodia transporter genes
Pharmacokinetics:
Chloroquine is generally administered orally, but in severe cases, like
falciparum malaria, may be treated by IV infusion or frequent small doses
IM or SC. it is completely absorbed from GIT, extensively distributed
throughout the tissues and concentrated in parasitised RBCS. Release from
tissues and infected erythrocytes is slow. The drug is metabolised in the
liver and excreted in the urine, (70% unchanged). Elimination is slow, halflife of 50 hours, and a residue persists for weeks or months.
Adverse effects
•
•
Nausea and vomiting, dizziness and blurring of vision, headach, and
urticaria are the most common unwanted effects. Bolus intravenous
injections can cause cardiac dysrhythmias.
Chloroquine is considered to be safe for use by pregnant women.
(2) 8-aminoquinoline derivatives
Primaquine the only 8-aminoquinoline dr. in clinical use.
Etaquine and tafenoquine are more active primiqune analogues with
prolonged duration and less side effects are still in the pre-clinical
trials.
Primaquine and its related drugs are active against the liver forms of
the parasite (tissue schizonticides), and they can effect a radical cure
of those forms of malaria in which the parasites have a dormant
stage (hypnozoites) in the liver(P. vivax and P. ovale).
Primaquine and its related drugs are active against the liver forms of
the parasite (tissue schizonticides), and they can effect a radical cure
and prevent relapse of those forms of malaria in which the parasites
have a dormant stage (hypnozoites) in the liver(P. vivax and P. ovale)
if given following to the treatment with a suitable blood
schizontocide.
Primaquine does not affect sporozoites and erythrocytic stage of the
parasite. However, it has a gametocidal action and is the most
effective antimalarial drug for preventing transmission of the disease
in all four species of plasmodia (causal prophylactic).
It is almost invariably used in combination with another drug, usually
chloroquine. Resistance to primaquine is rare.
Mechanism of action
• The exact MOA of 8-aminoquinoline drs. is unclear, but their
schizonticidal action may be due to generation of reactive oxygen
species or by interfering with the electron transport in the parasite.
• Also, Primaquine may bind to and alter the properties of protozoal
DNA.
Pharmacokinetics:
Primaquine is well absorbed on oral administration and is not concentrated
in tissues. It is rapidly metabolized to many compounds, the major one
being the deaminated drug. very little drug is present in the body after 1012 hours. The half-life is 3-6 hours. Tafenoquine is broken down much more
slowly and therefore has the advantage that it can be given on a weekly
basis. Metabolites are excreted in the urine
Adverse effects
1. Primaquine has few unwanted effects in most patients, but some
symptoms like GIT upset headache, visual disturbances and itching.
Large doses may cause methaemoglobinaemia with cyanosis.
2. Primaquine-associted methaemoglobinaemia is always less sever
and self-limiting in most cases.
3. Dangerous levels of methemoglobinemia only occur in patients G-6PD deficiency (people of African or Mediterranean descent). The
oxidant metabolic derivatives of primaquine. As a consequence, the
metabolic functions of the red cells are impaired and haemolysis
occurs.
4. Primaquine should not be administered to anyone with G-6-PD
deficiency because there can be a severe reaction with hemolytic
anemia.
5. Primaquine is contraindicated in pregnancy.
(3) 4-methanolquinolines
•
•
Quinine, Quinidine and Mefloquine are examples of this group of antimalarials.
Quinine and Quinidine are alkaloids derived from cinchona bark, while
Mefloquine is a synthetic analogue of quinin. All are blood schizonticidal drugs
effective against the erythrocytic forms of all four species of plasmodium. they
•
have no effect on preerythrocytic and exoerythrocytic forms or on the
gametocytes of parasite.
Their antiparasitic action is associated with; inhibition of the plasmodial haem
polymerase.
• With the emergence and spread of chloroquine resistance, quinine is
now the main chemotherapeutic agent for P. falciparum.
• Other pharmacological actions on host tissue include a depressant
action on the heart, a mild oxytocic effect on the uterus in pregnancy,
a slight blocking action on the neuromuscular junction and a weak
antipyretic effect.
• Quinidine has antiarrhythmic effect
a- Quinine and Quinidine
Pharmacokinetics:
Quinine is usually given orally and it is well absorbed, but it can also be
given by slow IV infusion for severe P. falciparum infections and in patients
who are vomiting. Quinidine is given only by parentral route.
The half-life of the drug is 10 hours; it is metabolised in the liver and the
metabolites are excreted in the urine within about 24 hours. Alkalinization
of the urine decreases its excretion.
Adverse effects
• Quinine has a bitter taste and is irritant to the gastric mucosa.
• The major adverse effect of quinine is cinchonism’ that characterized
by nausea, vomiting, dizziness, tinnitus, headache and blurring of
vision. Excessive plasma levels of quinine can result in hypotension,
cardiac dysrhythmias and severe CNS disturbances such as delirium
and coma. These effects are observed to some extent in all patients
receiving treatment and typically resolve with cessation of the
medication.
• Quinine can stimulate insulin release. Patients may have
hypoglycemia.
• Less commonly, hypersensitivity reaction with bronchospasm and
cutaneous manifestations, such as flushing and urticaria, may occur.
Mostly given in combination with other blood schizonticidal drugs like
pyrimethamine, dapsone or sulfadoxine. And antibiotic (eg, tetracycline,
doxycycline or clindamycin.
b- Mefloquine
Pharmacokinetics:
• Mefloquine is given orally and is rapidly absorbed. It has a slow onset
of action and a very long plasma half-life (up to 4weeks), which may
be the result of enterohepatic cycling or tissue storage.
• Mefloquine is metabolized primarily through the liver. Execretion
mainly in bile 95% and urine 5%. Elimination of mefloquine in anyone
with impaired liver function may be prolonged, resulting in higher
plasma levels and an increased risk of adverse reactions.
Adverse effects
• GIT disturbances,minimal cardiac arrythmia and transient CNS
symptoms like dizziness, confusion, dysphoria and insomnia can
occur.
• The drug is contraindicated to be used in those with a previous
history of seizures or a recent history of psychiatric disorders
including anxiety, hallucinations, depression, and suicidal ideations.
• Mefloquine users may experience sleep disturbances, including
strange dreams and insomnia.
• Mefloquine-induced pneumonitis is an infrequently reported but
serious adverse event in the setting of both prophylactic and
therapeutic use. One-third of the patients improved following
treatment with corticosteroids, and most patients fully recovered
upon discontinuation of the drug.
• Little is known about mefloquine teratogenic potential, so the WHO
approved the use of drug in the 2nd and 3rd trimester of pregnancy.
Women should however not become pregnant and should use
effective birth control while taking mefloquine and within 3 months
of stopping the drug, because of its long half-life and uncertainty
about its teratogenic effects.It may be used during breastfeeding,
though the drug appears in breast milk in low concentrations.
The combination of mefloquine plus artesunate appears to be effective
in most regions.
(4) Phenanthrene-methanols
•
•
Halofantrine and Lumefantrine are blood schizonticidal drug active
against all species of malarial parasite, including multidrug-resistant
strains of P. falciparum that are resistant to many antimalarials like
chloroquine, quinine, and pyrimethamine particularly when combined
with artemisinin derivatives.
In vitro studies suggest that halofantrine and Lumefantrine bind to and
inactivate plasmepesin, a haemoglobin degrading enzyme system unique
to the malarial parasites.
Pharmacokinetics:
Halofantrine is given orally. It is slowly absorbed, with a peak plasma
concentration achieved approximately 4-6 hours after ingestion (although
absorption is substantially increased by postprandial administration
especially with high fat-contents meal, It should be taken on an empty
stomach). The half-life is 1-2 days, although its main metabolite
(desbutylhalofantrine), which is equipotent to parent compound, has a
half-life of 3-5 days. Elimination is in the faeces.
Adverse effects:
1. Abdominal pain, GIT disturbances, headache, skin rashes , transient
increase in liver enzymes and cough may occur.
2. Halofantrine can produce cardiac arrhythmia (produce QT prolongation).
The drug should be avoided in patients with a history of dysrhythmia. It
should not be combined with another antimalarial that affect cardiac
conduction like mefloquine and quinine.
3. Because of such cardiotoxicity, halofantrine use is now limited to treat
infections caused by resistant organisms.
4. Some drugs like ketoconazole may decrease the drug bioavailability due
to inhibition of CYP3A4 activity.
5. It should be avoided in pregnancy.
(5) Antfolates
a. Pyrimethamine
- It is a 2,4-diaminopyrimidine (similar in structure to trimethoprim).
- It acts by interfering with DNA and RNA synthesis trough disturbing
tetrahydrofolic acid synthesis, by inhibiting the enzyme DHFR. It has a
greater affinity for the plasmodial enzyme than for the human
enzyme.
- It is active against the erythrocytic forms of the parasite.
- Pyrimethamine is given orally and is well, absorbed. It has a plasma
half-life of 4 days, and effective 'suppressive' plasma concentrations
may last for 14 days; it is taken once a week.
- Allergic reactions (rashes & hives), blood changes (leukopenia,
thrombocytopenia & hemolytic anemia) and hematourea are the
most common unwanted effects associated with pyrimethamine
therapy.
- In high doses, it may inhibit mammalian DHFR and cause a
megaloblastic anaemia ; folic acid supplements should be given if
this drug is used during pregnancy.
- Resistance the drug arises from single-point mutations in the genes
encoding parasite DHFR.
- Pyrimethamine is given orally and is well, absorbed. It has a plasma halflife of 4 days, and effective 'suppressive' plasma concentrations may last
for 14 days; it is taken once a week.
- Pyrimethamine is used only in combination with either Dapsone or a
Sulfonamide.
- Dapson is a sulfone drug mostly used in combination with
pyrimethamine in the treatment of malaria.
- Sulfadoxine is an ultra-long-lasting sulfonamide often used in
combination with pyrimethamine to treat or prevent
- The combination is considered to be more effective in treating malaria
caused by multiresistant P. falciparum
- Also, combination is indicated in the absence of a species-specific
diagnosis.
- Due to side effects associated with ptremethamine combined therapy, it
is used only to treat serious malaria infections or to prevent them in
areas where other drugs may not work.
b. Proguanil
- It is chemically related to biguanides.
- Same like pyrimethamine, it interferes with nucleic acid synthesis.
- It is active against the erythrocytic forms of the parasite . Proguanil
also has an additional effect on the initial hepatic stage but not on the
hypnozoites of P. vivax and P. ovale.
- It is given orally. Proguanil is a prodrug and is metabolised in the liver to
its active form, cycloguanil, which is excreted mainly in the urine. It is
well absorbed from GIT and its half-life is ~16 hours. It must be taken
daily.
- Proguanil is usually taken in combination with another anti-malarial
drug such as chloroquine or atovaquone (e.g., in the drug Malarone) to
treat multidrug-resistant strains of P. falciparum and P. vivax.
- Proguanil has few unwanted effects ; GIT distress and mild
hypersensitivity effects, and mild anxiety.
(6) Artemisnin and related derivatives
- Artemisinin is naturally isolated compound from Artemisia annua herb.
Because artemisinin itself has poor bioavailability that limit its
effectiveness, semi-synthetic derivatives of artemisinin, including
Artesunate, artemether and artether, have been developed .
- These compounds possess the most rapid action of all current
antimalarial drugs. They are fast-acting blood schizonticide effective in
treating the acute attack of malaria (including multidrug-resistant
falciparum malaria).
- All artemisinins used today are prodrugs of the biologically active
metabolite dihydroartemisinin, which is concentrated in parasitised
RBCs.
- The detailed mechanism of action is not clear, but one theory states
that when the parasite that causes malaria infects a red blood cell, it
consumes hemoglobin within its digestive vacuole, liberating free
heme, an iron-porphyrin complex. The iron reduces the peroxide bonds
in artemisinin generating high-valent iron-oxo species, resulting in a
cascade of reactions that produce ROS which damage the parasite
leading to its death.
- Artemisinin and its derivatives are also potent anthelmintics especially
in treatment of the human blood flukes infections, schistosomiasis,
which are the second most prevalent parasitic infections, after malaria.
- Artemisinin and related compounds are tested as anticancer drugs.
- Use of the drug by itself as a monotherapy is clearly discouraged by the
WHO as there have been signs that malarial parasites are developing
resistance to the drug. Instead combined therapy including artemisinins
(like artemether and lumefantrine ) are the preferred treatment for
malaria.
Pharmacokinetics:
- Artemisinin can be given orally, IM or rectally by suppository.
Artemether and artether are given either orally or IM, and artesunate
IM or IV.
- They are rapidly absorbed and widely distributed, and are converted in
the liver to the active metabolite dihydroartemisinin.
- The half-life of artemisinin is about 4 hours, of artesunate 45 minutes
and of artemether 4-11 hours.
Adverse effects:
a. Few adverse effects have been reported with artrmisinin and related
compounds.
b. For example, transient bradycardia, decrease in blood neutrophil count,
and brief episodes of fever have been reported.
(7) Hydroxynaphthoquinone derivatives
Atovaquone:
- It is used for the treatment of malaria and can prevent its development.
- Atovaquone is usually used in combination with the antifolate drug
proguanil, because they act together to cause a synergistic antimalarial
effect. The mechanism underlying this effect is not known, but synergy is
specific for this particular pair of drugs.
- It acts mainly as ubiquinone inactive analogue to inhibit the parasite's
mitochondrial electron transport chain.
- Atovaquone is active against both tissue and erythrocytic parsitic
stages, allowing chemoprophylaxis to be discontinued only 1 week after
the end of exposure
Pharmacokinetics:
- Atovaquone is administered orally. Its bioavailability is low and irregular,
but absorption is increased by fatty food. The drug is heavily proteinbound and has a half-life of 2–3 days.
- Most of the drug is eliminated unchanged in the feces.
Adverse effects:
a. When combined with proguanil, atovaquone is highly effective and well
tolerated. Few side effects of such combination have been reported, but
abdominal pain, nausea and vomiting can occur.
b. Pregnant or breast-feeding women should not take atovaquone.
c. Resistance to atovaquone monotherapy is rapid and results from a
single point mutation in the mitochondrial gene encodes cytochrome b,
while resistance to combined treatment with atovaquone and proguanil
is less common.
(8) Antibiotics
• Tetracycline, doxycycline, and clindamycin target prokaryotic protein
synthesis. In malaria parasites, these drugs appear to target the apicoplast, a
vital organelle in plasmodial cell
•
Antimicrobials have relatively slow blood schizonticidal effects because they
exert their toxic effects in the subsequent cycle of cell division . They are
typically paired with fast-acting antimalarials (usually quinine and
chloroquine).
• Doxycycline has a longer half life than tetracycline so is used more commonly.
• Resistance has not been detected to tetracycline, doxycycline or clindamycin.
• Adverse effects are common with the tetracyclines and interfere with
adherence. GI discomfort and candidiasis superinfection are the most
frequent complaints.
• Doxycycline therapy also poses a risk of esophageal ulceration and
photosensitivity especially if utilized for prolonged time in causal prophylaxis.
• Tetracyclines should not be given to pregnant women or children less than 8
years old because of the risk of deposition in growing bones and teeth.
•
Clindamycin is the preferred alternative in these groups.