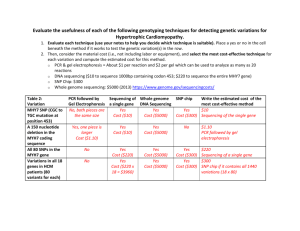
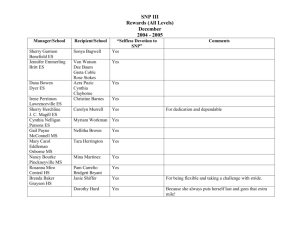
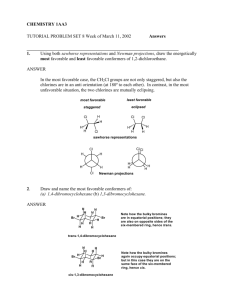
mec12003-sup-0003

Strategy for producing a SNP array in Atlantic Salmon
At the time this array was under development (late 2006), genomic resources for Atlantic salmon were relatively sparse forcing us to explore alternatives for SNP detection. In contrast to today's whole genome resequencing approaches, the majority of markers included on the SNP array were obtained after aligning and comparing sequences from either publically available EST sequences
(representing a range of individuals and localities), or re-sequencing of a reduced genome from a small number of commercial population fish.
Chip development and SNP discovery
Sequencing for SNP detection was performed in late 2006 and early 2007 using 454 technology.
Compared to today's output, the 454 platform was generating relatively little data per run, requiring us to pre-treat the genomic DNA and reduce the sample complexity. An enzymatic pretreatment was used, but in the absence of a reference genome it was not possible to predict in silico how much genome remained in the reduced fraction. Of the 6,927,968 reads generated, approximately 30% were eliminated due to repeat content, yet only 29% (1,404,933) of the remaining reads were included in contig bins, with the majority remaining as singletons. This indicates that the total complexity remaining in the material was too great to be captured with the sequencing performed and that additional sequencing or a more stringent sample reduction would have been helpful. At the individual sample level, variant detection was relaxed, with a minimum overlapping read requirement of 2, and minimum SNP coverage/total coverage ≥ 0.2. A consequence of this is that in contigs with low coverage, sequencing errors could be incorrectly assigned SNP status. However, the categorization of SNPs demanded that the variant base must appear in multiple samples before being accepted, reducing the possibility of false-positives due to sequencing error.