PROTEIN TURNOVER AND NITROGEN ECONOMY

PROTEIN TURNOVER AND NITROGEN ECONOMY
- proteins metabolism has a balance between body’s energy and synthetic needs
- dietary protein required to synthesize endogenous proteins (albumin, myosin, actin)
- essential amino acids cannot be synthesize by body; others can be synthesized from carbon sources
-table
- protein balance
relationship between synthesis and degradation (proteolysis) of proteins;
2.
roles of proteolysis : activation of enzymes (zymogens), blood clotting cascade, control of organ growth, digestion of dietary protein, fuel supply (starvation), maintain amino acid pools, regulate enzyme activity (removing enzyme from cell, half-lives), remove abnormal proteins, tissue repair
- starvation
glucose produced from amino acids (muscle proteins serves as fuel supply)
- to provide for proper balance during growth, proteolysis counterbalances synthesis to control organ size
- if dietary intake of amino acids > requirement for protein synthesis
new body protein synthesis ( positive balance) or body protein levels maintained at stable level ( neutral balance)
- positive nitrogen balance
occurs during growth when intake and storage of nitrogen exceed excretion of nitrogen; also associated with restoration of atrophied muscles or body building
- if protein intake is insufficient or if balance of amino acids ingested is incorrect for synthetic needs
endogenous protein catabolized to liberate free amino acids for synthesis of essential proteins ( negative nitrogen balance) ; associated with starvation and trauma; occurs when rate of proteolysis exceeds rate of protein synthesis (decrease rate of synthesis or accelerated digestion)
- elemental constituents of amino acids: carbon
CO
2
; hydrogen
H
2
O; nitrogen
urea or ammonia; sulfur
to SO
4
2-
1.
- insulin and glucocorticoids participate in regulation of protein turnover and nitrogen economy
- insulin
increase synthesis, decrease degradation of endogenous proteins ; favors maintenance of body protein pools; insulin-like growth factor (ILGF) promotes protein synthesis during growth
- glucocorticoids (released during stress or starvation)
peripheral tissue catabolism
- ala is a precursor for glucose synthesis; glucocorticoid catabolic effect coincides with ability of this class of hormones to promote gluconeogenesis
- insulin:glucocorticoid ratio determines net protein turnover ; fed state
high ratio
protein formation; fasting
insulin falls, low ratio
protein mobilized via proteolysis ; trauma
glucocorticoids increase, low ratio
protein mobilized via proteolysis
- endogenous protein degradation occurs in lysosome and cytoplasm ; membrane/extracellular proteins cycle through lysosome (proteolysis); lysosome is acidic; proteolysis in cytoplasm
(calpains, Ca
2+
dependent)
3. AMMONIA METABOLISM AND REMOVAL OF NITROGEN WASTE
Transamination reactions
- 1 st step in amino acid degradation is removal of amino nitrogen group by transferring it to alpha-ketoglutarate (alpha-KG) to produce glu ; catalyzed by aminotransferase/transaminases (cofactor is pyridoxal phosphate)
- pyridoxal phosphate derived from vitamin B
6
( also cofactor in glycogen phosphorylase and lysyl oxidase); deficiency
dermatitis, anemia, convulsions
- transaminases are reversible ; alpha-ketoacid accepts amino group from glu to produce new amino acid ; most common aminotransferases are for alanine (pyruvate) and aspartate
( oxaloacetate); aminotransferases test for liver damage
- transaminases transfer nitrogen to glutamate in non-hepatic tissues (muscle) to rid excess nitrogen from those tissues
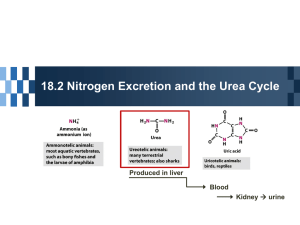
- in liver , nitrogen dumped onto glutamate as an initial step in conversion of nitrogen to excreted form
urea
Overview of nitrogen excretion
- body removes nitrogenous waste; some of these produces are from special starting materials while urea provides a means of removing nitrogen waste in a general manner
- kidney can excrete NH
4
+
as part of acidification mechanism of urine
- look at table
Nitrogen removal from nonhepatic tissues
- glutamate dehydrogenase ; one direction
reaction involves addition of nitrogen to alphaketoglutarate as ammonia ( non-hepatic tissues, remove harmful ammonia from these tissues)
- glutamate non transported across plasma membrane , but glutamine easily leaves cells
- glutamine formed through addition of a second ammonia molecule by glutamine synthetase to produce glutamine ; glutamine processed by kidney , which contains glutaminase
(with glutamate dehydrogenase) removes amino groups from glutamine resulting in alpha KG and ammonia ; ammonia released in this manner excreted in urine
4. UREA CYCLE
- liver glutamate produced by transamination gives up its nitrogen as free ammonia via glutamate dehydrogenase for eventual synthesis of urea (excreted)
1.
Carbamoyl phosphate synthetase-I
- urea cycle in liver (kidney)
- provides means of ridding body of nitrogen waste as urea
- ammonia from amino acids by combined actions of transamination and glutamate dehydrogenase
- mitochondria
ammonia incorporated into carbamoyl phosphate via carbamoyl phosphate synthetase-I (CPS-1)
reaction product, carbamoyl phosphate , provides substrate for cycle; reaction requires one ATP molecule providing phosphate that combines with CO
2 and ammonia and the other ATP molecule provides driving force for reaction (2
ATP)
- carbamoyl phosphate directly introduces the first source of nitrogen for the cycle
- CPS-1 is allosterically activated by N-acetylglutamate (produced by enzyme-catalyzed reaction of acetyl CoA + glutamate
N-acetylglutamate + CoA)
- mitochondrial CPS-1 (CPS1: NH
3
nitrogen source) distinguished from cytoplasmic CPS-2
(CPS-2: glutamine nitrogen source) in that CPS-2 is involved with pyrimidine synthesis
2. Ornithine transcarbamoylase
- first reaction of urea cycle: carbamoyl phosphate combines with ornithine
citrulline via ornithine transcarbamoylase ( occurs in mitochondrial matrix)
- ornithine transported into mitochondria form cytoplasm
- citrulline product released from mitochondria to cytoplasm in exchange for ornithine
3. Arginosuccinate synthetase
- cytoplasm
citrulline reacts with aspartate via arginosuccinate synthetase yielding arginosuccinate
- aspartate formed by transamination of glutamate with oxaloacetate
- aspartate is 2 nd
direct source of nitrogen for the cycle
- energy requiring reaction that cleaves ATP
AMP + PPi (costs two high-energy P bonds, PPi splits spontaneously into two Pi )
4. Arginosuccinase
- arginosuccinate cleaved by arginosuccinase into fumarate and arginine
- fumarate reconverted to oxaloacetate in citric acid cycle
can regenerate aspartate ; carbons from aspartate recycled with only nitrogen claimed for urea cycle
5. Arginase
- arginine cleaved to urea and ornithine ( into cytoplasm in exchange for citrulline)
- urea secreted by liver into blood to be cleared by kidney
- when arginase cannot handle accumulation of arginine
arginine stimulates formation of N acetylglutamate to increase formation of carbamoyl phosphate
reacts with ornithine to produce a mass action effect on arginase reaction thus increasing formation of urea
5. HYPERAMMONEMIA
Acquired hyperammonemia
- results from collateral circulation of portal system in response to liver damage (cirrhosis); blood flow from intestines bypasses liver
- collateral circulation (non cirrhosis) responsible for hyperammonemia; microorganisms in GI tract produce large amount of ammonia absorbed in portal system and sent to liver for detox ; portal-systemic shunting
blood flows directly to IVC (bypasses liver)
portal-systemic encephalopathy (PSE)
- shunting results in reduction of ammonia detoxification by liver ; ammonia from amino acid/protein metabolism cannot be converted to urea to an extent causing blood ammonia to rise
- liver transplant; reduce absorption of ammonia using lactulose
fermented by microorganisms to short chain organic acids that lower pH of intestinal lumen : converts NH
3
to
NH
4
+ ( not readily absorbed across intestinal epithelium and is excreted in stool)
Inherited hyperammonemia
- caused by deficiencies of urea cycle enzymes
- severity depends on proximity of defect to point of entry of ammonia in its processing to urea
- CPS-1 defects or ornithine transcarbamoylase defects
severe hyperammonemia ; these two defects can be distinguished by evaluating appearance of pyrimidines in urine; defect in ornithine transcarbamoylase
CPS-1 accumulates in mitochondria
excess carbamoyl phosphate leaks in to cytoplasm
increases rate of pyrimidine synthesis
- X-linked, in males
- high ammonia leads to mental retardation; possible reasons for neurologic damage:
1. ammonia reacts with alpha-ketoglutarate to form glutamate thus interfering with ATP production in citric acid cycle
2. excess glutamate formed undergoes amination to glutamine and then to alphaketoglutaramic acid , a neurotoxic compound
3. high ammonia
increase blood levels of some amino acids ; these compete with other amino acids for transport across blood-brain barrier ; thus, predominant transport of one or a few amino acids limits availability of other amino acids within the brain
reduction in normal rate of protein synthesis
Treatment
- restrict dietary protein
reduce amount of ammonia that must be detoxified
- alternative ammonia excretion mechanisms use body’s detox of exogenous chemicals
- benzoic acid
conjugated with glycine to form hippuric acid
readily excreted in urine taking with it the nitrogen from glycine; glycine is synthesized from CO
2 and NH
3
- phenylacetic acid
conjugated with glutamine forming phenylacetylglutamine
excreted in urine taking two nitrogens per molecule; glutamine continually synthesized in hyperammonemia in peripheral tissues (muscle) via glutamate dehydrogenase and glutamine synthetase reactions