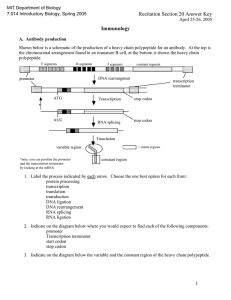
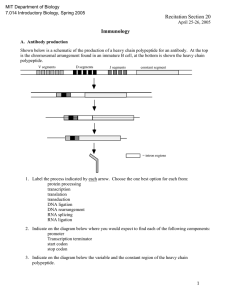
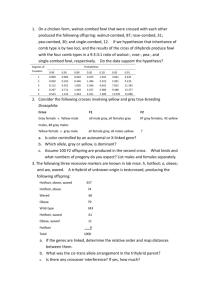
SubCav - Tool for sub-pocket comparison and alignment
advertisement

Kalliokoski T, Olsson TSG, Vulpetti A. J. Chem. Inf. Model. 2013, 53, 131-141. SubCav - Tool for subpocket comparison and alignment Dr. Tuomo Kalliokoski Lead Discovery Center GmbH, Dortmund, Germany Work conducted at Novartis Institutes for Biomedical Research, Basel, Switzerland 1972 1973 1974 1975 1976 1977 1978 1979 1980 1981 1982 1983 1984 1985 1986 1987 1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 Protein Databank (PDB) is growing • Number of searchable structures 1972-Mar 2013 100000 90000 80000 70000 60000 50000 40000 30000 20000 10000 0 How many fragments are there? 8 million unique chemical structures 2 million lead-like structures 400,000 Rule-Of-Three compliant structures Zuegg and Cooper. Drug-Likeness and Increased Hydrophobicity of Commercially Available Compound Libraries for Drug Screening. Curr Top Med Chem 2012, 12, 1500-1513. Bridging “Structural”-Space and “Fragment”-Space The information content of PDB is increasing Fragment chemical space is too large for experimental Fragment-Based Drug Design (FBDD) The need to develop tools for FBDD to take advantage of PDB! Binding site similarity “The availability of such data provides a basis for the identification of bioisosteres that are target specific. The resulting bioisosteres might be expected to provide more reliable information when modifying an existing lead compound than do existing approaches, which are based either on empirical measures of inter-substituent similarity or on non-target specific crystallographic data.” Kennewell EA, Willett P, Ducrot P, Luttmann C. Identification of target-specific bioisosteric fragments from ligand–protein crystallographic data. J Comput Aided Mol Des 2006, 20, 385-394. Subpockets and fragments BRICS* *Degen J, Wegscheid-Gerlach C, Zaliani A, Rarey M. On the art of compiling and using ’drug-like’ chemical fragment spaces. ChemMedChem 2008, 3, 1503–1507. SubCav • Tool for subpocket similarity searching and alignment • Based on pharmacophoric fingerprints with geometric hashing-inspired alignment • Source code available via anna.vulpetti@novartis.com Fingerprint descriptor SubCav atom type PDB atom types Acceptors with sp2 character (πacceptor) (A=) ALA.O ARG.O ASN.O ASN.OD1 ASP.O ASP.OD1 ASP.OD2 CYS.O GLN.O GLN.OE1 GLU.O GLU.OE1 GLU.OE2 GLY.O HIS.O ILE.O LEU.O LYS.O MET.O PHE.O PRO.O SER.O THR.O TRP.O TYR.O VAL.O α-carbon (CA) ALA.CA ARG.CA ASN.CA ASP.CA CYS.CA GLN.CA GLU.CA GLY.CA HIS.CA ILE.CA LEU.CA LYS.CA MET.CA PHE.CA PRO.CA SER.CA THR.CA TRP.CA TYR.CA VAL.CA LYS.NZ Donor (D) Donors with sp2 character (π-donor) (D=) ALA.N ARG.N ARG.NE ARG.NH1 ARG.NH2 ASN.N ASN.ND2 ASP.N CYS.N GLN.N GLN.NE2 GLU.N GLY.N HIS.N ILE.N LEU.N LYS.N MET.N PHE.N SER.N THR.N TRP.N TRP.NE1 TYR.N VAL.N Hydrophobe (H) ALA.CB ARG.CB ARG.CD ARG.CG ASN.CB ASP.CB CYS.CB CYS.SG GLN.CB GLN.CG GLU.CB GLU.CG HIS.CB HIS.CG ILE.CB ILE.CD1 ILE.CG1 ILE.CG2 LEU.CB LEU.CD1 LEU.CD2 LEU.CG LYS.CB LYS.CD LYS.CE LYS.CG MET.CB MET.CE MET.CG MET.SD PHE.CB PRO.CB PRO.CD PRO.CG SER.CB THR.CB THR.CG2 TRP.CB TYR.CB VAL.CB VAL.CG1 VAL.CG2 π-hydrophobe (H=) HIS.CD2 HIS.CE1 PHE.CD1 PHE.CD2 PHE.CE1 PHE.CE2 PHE.CG PHE.CZ TRP.CD1 TRP.CD2 TRP.CE2 TRP.CE3 TRP.CG TRP.CH2 TRP.CZ2 TRP.CZ3 TYR.CD1 TYR.CD2 TYR.CE1 TYR.CE2 TYR.CG TYR.CZ CA 9.3Å=4 D= 6.0Å=2 3.4Å=1 A= Bin Range (Å) 1 2.1-4.5 neutral donor & acceptor (P) HIS.ND1 HIS.NE2 SER.OG THR.OG1 TYR.OH 2 4.5-6.3 Ignored PRO.N and all HETATM 3 6.3-8.0 4 8.0-10.0 Alignment algorithm Implementation details Validation study • Align pairwise all similar subpockets in PSMDB* (non-redundant subset of PDB) • 3,268,620 pairs from 3,886 PDBs with 17,044 subpockets with 332 different fragments • Two alignment methods: – Fragment-based alignment – SubCav-based alignment * Wallach I, Lilien R. The Protein–Small-Molecule Database (PSMDB), A Non-Redundant Structural Resource for the Analysis of Protein-Ligand Binding, Bioinformatics 2009, 25, 615-620. When are two subpockets similar? • Two subpockets are similar if both after alignment have – Root-Median-Square-Deviation (RMSD) of fragments found in subpockets is less than 1.5 Å – Enough matched features* RMSD = 1.00 Overlap = 0.79 *Matched feature=if two features from the two subpockets are within 1 Å distance Very rarely subpockets with same fragments are geometrically similar... 3500000 3000000 2500000 2000000 Fragment-based OK SubCav- based OK Both OK 1500000 Not matched 1000000 500000 0 0.5 0.6 0.7 0.8 0.9 1 SubCav finds 73%-85% of fragmentbased (plus something else!) 120000 100000 80000 Fragment-based OK SubCav- based OK Both OK 60000 40000 20000 0 0.5 0.6 0.7 0.8 0.9 1 Three structures of thrombin aligned. The query (magenta) fragment-aligned (green) vs. SubCav aligned (cyan) Bioisosteric replacement example ACP Heat Shock Protein 90 (HSP 90) Bioisosteric replacement example Escherichia coli DNA gyrase B (sequence similarity 30%) Bioisosteric replacement example Adenine -> pyrazole? Escherichia coli DNA gyrase B (sequence similarity 30%) Bioisosteric replacement example HSP90 inhibitor Analysis of Histone Methyl-Transferase Binding Sites S-adenosylmethionine (SAM) or S-adenosyl-l-homocysteine (SAH) Fragmented in three: adenine, ribose, and tail fragments Pairwise SubCav-alignment and hierarchical clustering based on Overlap Analysis of Histone Methyl-Transferase Binding Sites The clustering of the cofactor binding site by subpockets around each specific fragment revealed different levels of local similarity within the selected proteins set. Analysis of Histone Methyl-Transferase Binding Sites The clustering of the cofactor binding site by subpockets around each specific fragment revealed different levels of local similarity within the selected proteins set. Analysis of Histone Methyl-Transferase Binding Sites A B C D Take home message Subpocket analysis can provide ideas in CADD Acknowledgements • Novartis Institutes for Biomedical Research: – Dr. Anna Vulpetti (mentor & co-author) – Education office (Presidential Postdoctoral Fellowship) • Cambridge Crystallographic Data Centre: – Dr. Tjelvar Olsson (mentor & co-author) • Chemical Computing Group: – Dr. Guido Kirsten (idea for alignment protocol)