Medical Devices
นายแพทย์ สหพล อนันต์ นาเจริญ
วิทยาลัยแพทยศาสตร์ พระมงกุฎเกล้ า
safeguarding the rights, safety, and wellbeing of the research subjects
Two primary protections people have for subject
of research are
1. Right risk benefit assessment
scientific/social value?
standard of care?
2. Valid informed consent
information
comprehension
voluntariness
Medical Devices/Drug
Drug: If the primary intended use of the product is
achieved through chemical action or by being
metabolized by the body
Device: does not achieve any of it's primary
intended purposes through chemical action and
which is not dependent upon being metabolized
for the achievement of any of its primary
intended purposes
Is The Product A Medical Device?
Medical Device Definition
• an instrument, apparatus, implement, machine,
contrivance, implant, in vitro reagent, or other similar or
related article, including a component part, or accessory
which is:
* recognized in the official National Formulary, or the
United States Pharmacopoeia, or any supplement to
them,
* intended for use in the diagnosis of disease or other
conditions, or in the cure, mitigation, treatment, or
prevention of disease, in man or other animals, or
* which does not achieve any of it's primary
intended purposes through chemical
action within or on the body of man or
other animals.
* which is not dependent upon being
metabolized for the achievement of any of
its primary intended purposes.
• GeneSearch™ BLN Test Kit - P060017
metastases larger than 0.2 mm in nodal tissue removed
from biopsies of breast cancer patients.
• Binax Now® Malaria Test - K061542
plasmodium parasites using a whole blood sample
drawn from a vein or obtained by a finger stick.
• LightTouch™ Non-invasive Cervical Cancer
Detection
• Access: FDA’s Center for Devices and
Radiological Health (CDRH) Classification
Database.
• Device Determination Officer, Office of
Compliance
การศึกษาความสาเร็จของการหย่าเครื่องช่ วยหายใจเปรียบเทียบระหว่ างการใช้ เครื่อง
SmartCare/PS กับ การใช้ T-piece weaning
computer-driven weaning protocol: difficult weaning
การศึกษาเพือ่ ประเมินความปลอดภัยและประสิ ทธิภาพของการใช้ ขดลวด
เคลือบยา….ในการรักษาผู้ป่วยหลอดเลือดหัวใจตีบ
การศึกษาความแม่ นยาในการตรวจวินิจฉัย Human Papilloma Virus
โดยวิธีตรวจหา DNA โดยใช้ Hybrid Capture II assay
เปรียบเทียบกับการตรวจทางพยาธิวทิ ยา
“safety and effectiveness of the new device”
Medical devices
• For marketing
Premarket Notification (510k)
510(k) Exempt Devices
Premarket Approval (PMA)
• For investigation
Investigational Device Exemption
(IDE): 21 CFR 812
Device Classification
• The Food and Drug Administration (FDA) has
established classifications
– 1,700 different generic types of devices
– grouped them into 16 medical specialties
Each of these generic types of devices is assigned to
one of three regulatory classes based on the level of
control necessary to assure the safety and
effectiveness of the device.
Device Classification
classification is risk based
• the risk the device poses to the patient
and/or the user is a major factor in the
class it is assigned.
• Class I includes devices with the lowest
risk
• Class III includes those with the greatest
risk.
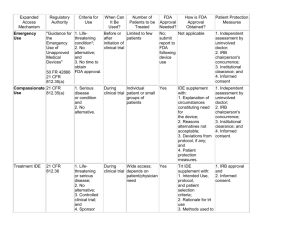
Investigational Device
Exemption (IDE)
• IDE: allows the investigational device to be
used in a clinical study in order to collect
safety and effectiveness data required to
support a Premarket Approval (PMA)
application or a Premarket Notification
[510(k)] submission to FDA.
IDE needed
• Significant risk device research
• Non-significant risk device research
– IRB approval; after which the study is considered
to have an IDE
– IRB disagrees and determines that the device poses
a significant risk, the sponsor must report this finding
to FDA within five working days
• Clinical investigations of devices where
safety and effectiveness data are collected
• New intended use of an approved device
Medical devices
IRB must know three basic things
1. Does the study involve a device?
2. Is the device SR or NSR?
3. Does it raise new questions of safety and
effectiveness?
Is the device SR or NSR?
• Significant risk device
Intended as an implant and
– presents a potential serious
risk to the health, safety, or welfare
of a subject
e.g. pace maker,
deep brain stimulators,
implanted fecal incontinence device
Implantable Middle Ear Hearing Device
• Is purported or represented to be for a use in
supporting or sustaining human life
– and presents a potential serious risk to the health,
safety, or welfare of a subject
e.g. respirators, dialysis machines
• Is for a use of substantial importance in
diagnosing, curing, mitigating, or treating
disease, or otherwise preventing impairment
of human health
– and presents a potential serious risk to the health, safety, or
welfare of a subject
• Otherwise presents a potential for serious
risk to the health safety or welfare of the
subject
– e.g. kit, dna detection, human papillomavirus, tissue adhesives
for use in neurosurgery, bronchial tube, sutures.
Does it raise new questions of
safety and effectiveness?
• Device classification depends on
– the intended use of the device and
– indications for use
Approved device: New intended use
New device: new materials or
design used in approved devices
New device: new indication
Does it raise new questions of
safety and effectiveness?: NO
1. Approved devices used in accordance with
labeling
2. Only using the device to address the research
question – not safety and effectiveness
– Stapled Hemorrhoidectomy versus
Conventional Excision Hemorrhoidectomy
for Acute Hemorrhoidal Crisis
3. Testing of a minor modification,
4. Testing of a combination of approved devices
Does it raise new questions of
safety and effectiveness?: NO
A diagnostic device if it complies with the labeling
requirements in §809.10(c) and if the testing:
a. is noninvasive;
b. does not require an invasive sampling
procedure that presents significant risk;
c. does not by design or intention introduce
energy into a subject; and
d. is not used as a diagnostic procedure
without confirmation by another medically
established diagnostic product or procedure;
Does it raise new questions of safety
and effectiveness?: YES
1. Clinical investigations of devices where safety and
effectiveness data are collected
2. Significant risk device research
3. New intended use of an approved device
Different age population
New disease or condition
Different body placement
Changing from ‘treatment’ to ‘prevention’ for
the same disease
4. Also, new materials or design used in approved devices
• All medical devices
– must be manufactured under a quality
assurance program,
– be suitable for the intended use,
– be adequately packaged and properly
labeled,
– have establishment registration and device
listing forms on file with the FDA.
Physician should
- use firm scientific
rationale and sound
medical evidence
-use in accordance with
labeling
-well informed about the
product
-monitoring of the study
-required records and
reports.
Marketing
Substantial Equivalence (SE)
• A 510(k) requires demonstration of
substantial equivalence to another legally
U.S. marketed device.
• Substantial equivalence (SE) means that
the new device is at least as safe and
effective as the predicate.
Device Class and Regulatory Controls
•
Class I General Controls
– 510(k) Exempt Devices
– Without Exemptions
•
Class II General Controls and Special
Controls
– 510(k) Exempt Devices
– Without Exemptions
•
(510k)
(510k)
Class III General Controls and Premarket
Approval
(PMA)
A device is substantially equivalent
if, in comparison to a predicate it:
– has the same intended use as the predicate;
and
– has the same technological characteristics as
the predicate;
or
A device is substantially equivalent
– has the same intended use as the predicate;
and
– has different technological characteristics and
the information submitted to FDA;
• does not raise new questions of safety and
effectiveness; and
• demonstrates that the device is at least as safe
and effective as the legally marketed device.
In order to conduct a significant risk device
study
• submit a complete IDE application to FDA
for review and obtain FDA approval of the
IDE;
• device must be manufactured under a
quality assurance program
• submit the investigational plan and report
of prior investigations to the IRB at each
institution where the investigation is to be
conducted for review and approval; and
• select qualified and obtain signed
investigator agreements.
• an IDE application is considered approved
30 days after it has been received by FDA,
unless FDA otherwise informs the sponsor
prior to 30 calendar days from the date of
receipt, that the IDE is approved,
approved with conditions, or disapproved.
• Labeling - The device must be labeled in
accordance with the labeling provisions of the
IDE regulation and must bear the statement
"CAUTION - Investigational Device. Limited by
Federal (or United States) law to investigational
use."
• Distribution - Investigational devices can only be
distributed to qualified investigators.
• Informed Consent - Each subject must be
provided with and sign an informed consent form
before being enrolled in the study. Protection of
Human Subjects, contains the requirements for
obtaining informed consent
• Monitoring - All investigations must be properly
monitored to protect the human subjects and
assure compliance with approved protocols
under
• Prohibitions - Commercialization,
promotion, and misrepresentation of an
investigational device and prolongation of
the study are prohibited
• Records and Reports - Sponsors and
investigators are required to maintain
specified records and make reports to
investigators, IRBs, and FDA
Premarket Approval
• 1. ครื่ องมือแพทย์ ทตี่ ้ องมีใบอนุญาต ได้แก่ ถุงยางอนามัย ถุงมือยาง
สาหรับการตรวจโรค ถุงมือยางสาหรับการศัลยกรรม กระบอกฉีดยาผ่าน
ผิวหนังปราศจากเชื้อชนิดใช้ครั้งเดียว กระบอกฉีดยาอินซูลนิ ปราศจาก
เชื้อชนิดใช้ครั้งเดียว ชุดตรวจการติดเชื้อเอชไอวีเพื่อวินิจฉัยโรค
Premarket Notification
• 2. เครื่องมือแพทย์ที่ต้องแจ้ งรายการละเอียด
เครื่ องมือแพทย์กลุ่มนี้ ผูป้ ระกอบการผลิต นาเข้า หรื อขายต้องแจ้งรายการละเอียด
ต่อเลขาธิ การคณะกรรมการอาหารและยา ตามกฎกระทรวง ฉบับที่ 4 (พ.ศ.
2533) เครื่ องมือแพทย์ในกลุ่มนี้ได้แก่
2.1 ชุดตรวจการติดเชื้อเอชไอวี เพื่อวัตถุประสงค์อื่น : ประกาศกระทรวง
สาธารณสุ ข ( ฉบับที่18 ) พ.ศ. 2538
2.2 เครื่ องใช้หรื อผลิตภัณฑ์ที่ใช้เพื่อกายภาพบาบัด : ประกาศกระทรวง
สาธารณสุ ข (ฉบับ ที่ 19) พ.ศ. 2539
2.3 เครื่ องตรวจวัดระดับหรื อปริ มาณแอลกอฮอล์ในร่ างกาย : ประกาศ
กระทรวงสาธารณสุ ข (ฉบับที่ 22) พ.ศ. 2540
2.4 เต้านมเทียมซิ ลิโคนใช้ฝังในร่ างกาย : ประกาศกระทรวงสาธารณสุ ข
(ฉบับที่ 23) พ.ศ. 2540
• 3. เครื่องมือแพทย์ นาเข้ าทั่วไป
เครื่ องมือแพทย์ที่ไม่จดั เข้าข่ายเครื่ องมือแพทย์ที่ตอ้ งมีใบอนุญาต หรื อที่
ต้องแจ้งรายการละเอียด จัดเป็ นเครื่ องมือแพทย์ทวั่ ไปที่ผผู ้ ลิต ผูน้ าเข้า
หรื อผูข้ าย ไม่ตอ้ งรับใบอนุญาตหรื อแจ้งรายการละเอียด แต่
ผูป้ ระกอบการนาเข้าต้องแสดงหนังสื อรับรองการขายเครื่ องมือแพทย์ใน
ประเทศผูผ้ ลิต ซึ่งออกโดยทางราชการหรื อ สถาบันเอกชนที่ทางราชการ
ของประเทศนั้นรับรองและ ผ่านการตรวจสอบจากคณะกรรมการอาหาร
และยาแล้ว แสดงตอพนั
กงานเจ้าหน้าที่ ณ ดาน
่
่
ศุลกากรตามประกาศ กระทรวงสาธารณสุ ข ฉบับที่ 34 (พ.ศ.
2549)
Investigational plan
(a)Purpose. The name and intended use of the device and
the objectives and duration of the investigation.
(b)Protocol. A written protocol describing the methodology
to be used and an analysis of the protocol demonstrating
that the investigation is scientifically sound.
(c)Risk analysis. A description and analysis of all increased
risks to which subjects will be exposed by the
investigation; the manner in which these risks will be
minimized; a justification for the investigation; and a
description of the patient population, including the
number, age, sex, and condition.
Investigational plan
(d)Description of device. A description of each
important component, ingredient, property, and
principle of operation of the device and of each
anticipated change in the device during the
course of the investigation.
(e)Monitoring procedures. The sponsor's written
procedures for monitoring the investigation and
the name and address of any monitor.
(f)Labeling. Copies of all labeling for the device.
Investigational plan
((g)Consent materials. Copies of all forms
and informational materials to be provided
to subjects to obtain informed consent.
(h) Additional records and reports. A
description of records and reports that will
be maintained on the investigation in
addition to those prescribed in subpart G.
Monitoring investigation
(a) Securing compliance
investigator is not complying with the signed agreement,
the investigational plan, the requirements of this part or
other applicable FDA regulations, or any conditions of
approval imposed by the reviewing IRB or FDA shall
promptly either secure compliance, or discontinue
shipments of the device to the investigator and terminate
the investigator's participation in the investigation. A
sponsor shall also require such an investigator to
dispose of or return the device, unless this action would
jeopardize the rights, safety, or welfare of a subject.
Monitoring investigation
(b)Unanticipated adverse device effects.
(1) A sponsor shall immediately conduct an evaluation of
any unanticipated adverse device effect.
(2) A sponsor who determines that an unanticipated
adverse device effect presents an unreasonable risk to
subjects shall terminate all investigations or parts of
investigations presenting that risk as soon as possible.
Termination shall occur not later than 5 working days
after the sponsor makes this determination and not later
than 15 working days after the sponsor first received
notice of the effect.