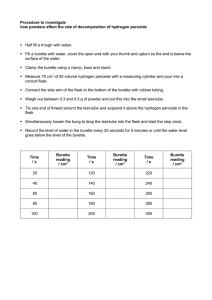
M. Sc. - Chemistry ( Part- I ) Department of Chemistry Pratap College Amalner Inorganic Chemistry Practical CH-I-1 The student should perform minimum of 18 experiments. 2) Draw Flow chart all experiments except for thermochemistry, Chromatography and instrumental method 3) Use of double method is compulsory for all volumetric analysis except instrumental method 1. Analysis of ore a) Pyrolusite ore - Estimation of silica gravimetrically and Manganese volumetrically. b) Haematite - Estimation of iron volumetrically and silica gravimetrically. 2. Analysis of binary mixtures by gravimetric and volumetric method a) Copper- Nickel b) Copper-Zinc c) Iron-Magnesium 3. Preparation of the following and its purity : a) Potassium tris(acetylacetonato)iron(III) b) Potassium di aqua bis(oxalato) chromate (III) c) Chloro penta-amminecobalt (III) chloride 4. Drug Analysis a) Determination of iron from given drug sample. 5. Thermochemistry (Any two salts) To determine the lattice energy of binary salts ( NaCl, KCl, CaCl2). 6 Chromatography a) Determination of the Rf value of Pb,Cu,Cd ions by using paper chromatographic technique. b) Determination of the Rf value of Fe, Al, Cr ions by using paper chromatographic technique. 8. Instrumental method of Analysis ( Five Experiment) A) To determine the strength of given mixture of carbonate and bicarbonate by pH metric method B) To determine the Ca in given solution flame photometrically, by calibration curve Method. C) Spectrophotometry a) Estimation of Manganese from steel. D) To determine the amount of copper present by iodometric method (potentiometrically) E) Estimation of Boric acid using NH4OH by conductometric method. Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim: Estimation of silica and manganese in pyrolusite. Theory :Pyrolusite ore contains a large amount of manganese in the form of manganese dioxide, MnO2, and little silica. It is a black mineral. Its manganese content is between 60 to 63 % and silica content is between l0 to l5% .The dissolution of the ore is done in the mixture of conc. HCl and conc.HNO3. The insoluble residue contains silica (SiO2) and soluble part contains largely manganese. Silica is estimated gravimetrically while manganese is estimated volumetrically, using KMnO4. Potassium permanganate is a powerful oxidizing agent in acidic medium and hence can be used to estimate Mn volumetrically. The method is called Volhard's method. Chemicals: conc. HCl , conc. HNO3 , conc. H2SO4 , Zinc oxide paste (emulsion) , KMnO4 solution (approx. 0.1 N) oxalic acid Apparatus : Stem-cut funnel, Silica crucible , 100 ml. Volumetric flask Procedure : Dissolution of the ore : 1. Weigh accurately about 0.3 to 0.4 g of the ore. call it as w g. Transfer it in a conical flask. 2. Add to it l0 ml. conc. HCl and 3 ml. conc. HNO3. cover it with a stem-cut funnel which actsas an air condenser. 3. Heat the flask in a fuming cupboard on a low flame till black particles of the ore dissolve. Ifthe ore does not dissolve, again add 2 ml. conc.HCI and 1 ml. conc. HNO 3 and heat it again. 4. Repeat the procedure till the ore dissolves. 5. Cool the flask and add 2 ml. conc. H2SO4 slowly from the sides of flask. 6. Heat the flask again till white copious fumes of SO3 start evolving. 7. Cool the solution and rinse the funnel and its stem with little distilled water in the same flask.Add to it about 20 ml. distilled water. 1. 2. 3. 4. Estimation of silica (SiO2) gravimetrically : Filter the above solution through whatman No. 41 filter paper collect the filtrate in a 50ml.beaker. Transfer all the silica in a filter paper. Wash it with hot water till the residue is free from chloride (test with AgNO3) and acid (test by blue litmus paper).Collect the filtrate and washings in the same beaker. Dilute it to exactly 100 ml in a volumetric flask. Preserve it for the volumetric estimation of Mn. Dry the filter paper on a metal cone at 110 to 1200C. Ignite the residue along with the filter paper in previously weighed silica crucible and heat itfor about 30-40 minutes. Cool the crucible and its lid. Weigh the crucible with lid and the residue. Find the weight of SiO2.Call as X g. Calculate the percentage of SiO2 in the ore.Repeat this operation till constant weight. Estimation of Mn yolumetrically (Volhards method) Principle: This method is based on the oxidation of divalent manganese with potassium permanganate in a hot (not less than 80°C) solution neutralised with zinc oxide and containing a little excess of the latter. The endpoint is indicated by the persistent pink colour. Solid zinc oxide have a tendency to adsorb manganese ion. Potassium permanganate was stable in boiling hot solution at pH 7.08 which was obtained by saturation with zinc oxide. Fading in pink of potassium permanganate observed near the end point of the titration During the dissolution of pyrorusite ore, MnO2 gets converted to MnSO4i,e, Mn (IV) gets converted to Mn (II). Volhard,s method is based on the principle that when manganous salt solution is warmed and titrated with standard KMnO 4 solution turns to brown and dark precipitate of hydrated manganesedioxide is obtained. During the reaction manganous ion is oxidized and permanganate ion is reduced giving hydrous manganese dioxide. 2KMnO4 + 3MnSO4 + 2H2O i.e. 3Mn2+ + 2MnO4- + 2H2O K2SO4+ 5MnO2 +2H2SO4 5MnO2 + 4H+ Hydrated manganese dioxide has acidic properties and absorbs manganous hydroxide Preventing complete oxidation. This difficulty is avoided by introducing sufficient amount of ZnO paste. Function of ZnO paste(emulsion): i) The precipitate hydrated of MnO2 settles very quickly. ii) It neutralisesH2SO4 which is added during the titration of manganese iii) It avoids formation of potassium permanganate. iv) It avoids formation of manganous acid, hence prevents precipitation of manganous manganite. v) It forms Zn ferrite with Fe if present as an impurity in MnSO 4. Thus interference of Fe is masked. Reactions: MnO2 + 4HC1 MnC12 + 2H2O + Cl2 MnO2 + 4HNO3 Mn(NO3)2 + 2H2O + 2NO2 +O2 MnCl2 + H2SO4 MnSO4 + 2HC1 Mn(NO3)2 + H2SO4 MnSO4 + 2HNO3 2KMnO4 + 3MnSO4 + 2H2O 5MnO2 + K2SO4 +2H2SO4 Atomic Wt.x 3 5 xchange in oxidation state ZnSO4 + H2O ZnO + H2SO4 Procedure: Part I -Preparation of standard 0.1N Oxalic acid solution (Eq. Wt.63) 1000 ml 1N oxalic acid solution contains 63 g of oxalic acid. 1000 ml 0.1N oxalic acid solution contain 6.3 g of oxalic acid. 250ml 0.1N oxalic acid solution contain 1.575 g of oxalic acid. Weigh accurately 1.575 g of oxalic acid (H2C2O4.2H2O) on a watch glass. Transfer it in a beaker and rinse the watch glass with little water in the same beaker. Dissolve it in about 50ml distilled water. Transfer it into 250 ml volumetric flask. Dilute it up to the mark using distilled water. Use of this solution for standardization KMnO4 solution. Part II :Standardisation of KMnO4solution : 1. Fill burette no.1 by 0.1 N (approx.)KMnO4 solution.Fill burette no.2 by 0.1 N oxalic acid Take by burette no.2, 10 ml of 0.1 N oxalic acid and add to it 15 ml 2N H2SO4solution 2. Heat the solution on a wire gauze to 70o C and titrate this hot solution with KMnO4 solution added from burette no.1. 3. The end point of the titration is noted when permanent faint pink colourappears.Call this 4. burette reading as X1 ml. 5. To the same solution, add one ml oxalic acid solution by burette no.2 and heat the flask to 6. 70oC.The solution becomes colorless. 7. To this hot solution, add KMnO4solution from burette no.1 till faint pink colour appears. 8. Call this burette reading as X2 ml. 9. To the same solution, add one ml oxalic acid solution by burette no. 2 and heat the flask to 70oC.The solution becomes colorless. 10. To this hot solution, add KMnO4 solution from burette no.1 till faint pink colour appears. 11. Call this burette reading as X3 ml. 12. From these three burette readings find out the exact normality of KMnO 4 solution. Part III : Estimation of Manganese : 1. Fill the burette no.1 by standardized KMnO4solution.and burette no.2 with diluted solution of MnSO4. 2. Take by burette no.2, 10 ml of the diluted solution of MnSO4. Add 1/2 test tube of ZnO paste. 3. Add one test tube water to it. 4. Heat the solution on water bath between 40 and 600C and add 2 to 3 drops of 2N HNO3 solution (which promotes the settling of the ppt. of MnO2 at the bottom). 5. Titrate this hot solution with standardised KMnO4 solution from burette no.1 with constant shaking.The end point is faint pink colour to supernatant liquid which persists even after vigorous shaking. Note down this burette reading . The faint pink colour should remain as it is even after heating, if not add one or two drops of KMnO4 solution. 6. Take two more readings. Find the constant burette reading as Y ml. Imp. Note : During the titration it is necessary allow the ppt. to settle down and see the colour of the supernatant liquid against white background after each reading. Observation and Observation Table – Standardisation of KMnO4 solution – Burette 1 : 0.1 N (approx) KMnO4 solution Burette 2 : 0.1 N exartH2C2O4 . 2H2O solution Indicator : KMnO4 act as a self indicator End Point : Colourless to pink Burette – 2 Burette – 1 10 ml X1 = 12 ml X2 = ml ml 14 ml X3 = ml Calculations:Find out exact normality of KMnO4 KMnO4 = Oxalic acid 0.1 x 10 X1 N1x X1 = 0.1 x 10 ∴N1 = N2 x X2 = 0.1 x 12 ∴N2 = 0.1 x 12 X2 N3 x X3 = 0.1 x 14 ∴N3 = 0.1 x 14 X3 Exact normality of KMnO4= mean N4 = N1 N 2 N 3 3 Estimation of Mn from a given solution – Observation and Observation Table – Burette 1 : 0.1 N (approx) KMnO4 solution Burette 2 : diluted Mn2+ solution Indicator : KMnO4 act as a self indicator End Point : Colourless to pink Burette – 2 10 ml 10 ml Burette – 1 Y1 = ml Y2 = ml Estimation of Manganese: (Volumetrically) 1000 ml 1 N KMnO4 = 16.462 g of Mn. Y ml N4 Normal KMnO4 = 10 ml dil. solution 100 ml dil. solution ? 100 of ore = A g of Mn = A g of Mn g of ore = ? % of Mn YxN 4 x16.46 1000 C.B.R Y=___ml = Z g of Mn. = Z g of Mn. = ? Wg 10 ml Y3 = ml Zx100 10 Ax100 W = ---- % Calculations: Estimation of Silica (Gravimetrically) Weight of ore sample Weight of empty crucible Weight of empty crucible + Residue Weight of residue W g of the ore sample contains 100 g of the ore sample contains 100x X W = W g = W1 g = W2 g = W2-W1= X g = X g of SiO2. = ? = ------ % of SiO2 Result Table: Sr.No. 1. 2. 3. 4. 5. 6. Description Weight of Pyrolusite taken Amount of Silica in sample % of SiO2 Exact Normality of KMnO4 Amount of Mn in sample % of MnO2 Values Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim :- Estimation of silica and iron in hematite ore Principle:Haematite ore contains mainly iron oxide Fe2O3 With impurities such as Al2O3 and SiO2 Chemicals :- Conc. HCl ,Conc. HNO3 , Perchloric acid 60% , K3Fe(CN)6 , K2Cr2O7 (0.025N) ,SnCl2 sol.(4%) ,HgCl2 sol.(saturated). Procedure: The dissolution of Ore: About 0.25 g of ore is accurately weighed in 250 ml conical flask add to it 15 ml con. HCl. Place a stem-cut funnel in the mouth of the flask. Heat the flask till the ore is completely dissolved. Cool and add to it con. HNO3 about 5 ml and heat for 10 minutes. Cool thoroughly and add 5 ml 60% perchloric acid and heat till white fumes starts evolving. Cool the flask. Rinse the funnel with water and dilute the contents to about 100 ml. with distilled water. Estimation of Silica: Filter the above solution through Whattman filter paper No. 41. Pour and wash the residue with hot water till it is free from acid as tested with litmus paper. Collect the Filtrate and washing. Dilute-it to 250 ml and reuse for estimation of iron (solution B). Dry and ignite the residue. Weigh silica in previously weighed crucible and calculate the % of silica. Estimation of Iron volumetrically Pipette out 100 ml diluted solution B in 500 ml beaker add 3 ml conc. HNO 3, 7.5 g of ammonium Chloride, 5 to 6 drops of bromocresol purple indicator and heat to boiling and add slowly to the above solution 1:1 ammonia till iron is completely precipitated. (i.e. supernent liquid turns purple). Filter through ordinary filter paper and wash with hot water three times. Dissolve the precipitate in hot 1:1 HCl (About 10 ml) and wash the filter paper with hot water till free from acid. Dilute the filtrate to 100 ml in volumetric flask. Fill it in burette 2. Take out 10 ml of above solution, heat to boiling and add to it 4% SnCl2 solution drop wise till becomes colourless, add a drop or two drops SnCl2 in excess to ensure the complete reduction. Cool thoroughly and add solution of HgCl2 till slight turbidity is obtained. Finally titrate with standard K2Cr2O7 (0.025 N) using potassium ferricyanide solution as an external indicator. Calculate % of iron present in Ore. Iron is estimated using K 2Cr2O7 solution by two methods: A. External Indicator: K3Fe(CN)6 Potassium ferricynide. Fill burette no.1 with diluted solution of iron and burette no.2 with 0.025N K2Cr2O7 solution. Take by burette no.1, 9 ml. of diluted iron solution in a100 ml. conical flask. 1. Add to it 2 ml. conc. HCl and boil it. It becomes yellow in colour. 2. To this hot solution add drop by drop 4% SnCl2 in excess to ensure complete reduction of ferric iron to ferrous iron. 3. Cool the solution in the flask thoroughly under tap water. 4. To this cooled solution add drop wise saturated solution of HgCl2 till a slight turbidity is just obtained. (Caution: Avoid excess of HgCl2 otherwise it will oxidize Fe2+ to Fe3+). 5. Titrate this solution with 0.025N K2Cr2O7 using K3 [Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X1 ml. 6. To the same solution, add 1 ml of iron solution from burette no.1. 7. Add 2-3 drops of conc. HCl and boil. 8. To this hot solution add carefully drop by drop SnCl2 solution .Add one or two drops o f HgCl2 solution till a slight turbidity is just obtained. 9. Titrate it with 0.025N K2Cr2O7 solution using K3[Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X2 ml. In a similar way take one more reading. Call this reading as X 3 ml. 10. B. Internal indicator: Diphenyl amine. Potassium dichromate acts as an oxidizing agent in acidic medium and oxidizes Fe (II) to Fe (III) ions. Diphenylamine is colorless, so long as K2Cr2O7 is completely used up in the reaction. At the end point, a slight excess of K2Cr2O7 oxidizes the indicator to intense violet colour. Reactions: Fe2O3 + 6HCl 2FeCl3 + 3H2O 2FeCl3 + SnCl2 2FeCl2+ SnCl4 SnCl2+ 2HgCl2 SnCl4 +Hg2Cl2 SnCl2+ HgCl2 SnCl4 +Hg (Gray ppt.) 6FeCl2+14HCl+K2Cr2O7 6FeCl3+2KCl+2CrCl3 +7H2O Calculations: 1. % of silica in Ore 0.25g of Ore 100 g of Ore 100 X x = x g of SiO2 = --- g of SiO2 0.25 = ------% of SiO2. 2. % of Iron in Ore. Observations: a) Burette 2- 0.025 K2Cr2O7 solution b) Burette 1 solution – Diluted solution c) Indicator – K3[Fe (CN) 6] d) End point – Sr. No. 1. 2. 3. Burette 1 Solution (in ml) 10 10 10 Burette 2 reading (in ml) Constant Burette reading (in ml) X ml Calculations: 1000 ml 1N K2Cr2O7 = 55.85 g of Iron. =? X ml N1 Normal K2Cr2O7 10 ml solution = A g of Iron 100 =? ml solution 250 ml solution =? 0.25 g ore sample contains = C g of Iron =? 100 g of sample % of Iron in Ore simple Result: 1) % of Silica in Ore sample 2) % of Iron in Ore sample = -------% Fe = = XxN 1 x55.85 1000 100 xA 10 250 xB 100 100 xC 0.25 = A g of Iron = B g of Iron = C g of Iron Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_________________________ Batch:____________ Aim : Separation of Cu-Ni mixture and estimate the amount of Ni gravimetric and Cu volumetric methods from the given binary mixture solution Theory: Binary mixture contain copper and nickel, in which copper is Group II radical and Nickel is group IIIB radical. According to qualitative analysis technique separation of group II is done from the mixture.By adding dilute HCl and passing H2S gas copper get separated as CuS precipitate and filtrate is use for estimation of nickel . Requirements : Given binary mixture of Cu-Ni; Whatman paper No. 41, ethyl alcohol, 0.01M EDTA solution, 0.01M ZnSO4, 1% DMG, conc. HCl,1:1 NH3, EBT indicator,pH 10 buffer( Not ammonia buffer), Fast sulphone black F indicator Procedure : (A) Separtion of Cu-Ni mixture: 1. Dilute the given solution of mixture of Cu and Ni to 100 ml with distilled water in a standard measuring flask. Shake the flask well. Transfer it in a 250 ml beaker. 2. Add 20-25 ml dilute HCl ( two test tube) in it and warm the solution, then pass H 2S gas through it till copper is completely precipitated. 3. Filter the black precipitate of CuS through ordinary filter paper .Use precipitate for estimation of copper and filtrate for estimation of nickel. (B) Estimate the amount of Ni gravimetric:1. Collect all the filtrate along with washing solution in 250ml beaker and add 1 ml conc. HCl, and boil it to expel H2S. 2. To this hot solution add drop wise 30 ml of 1% DMG solution. 3. Then add 1:1 NH3 till drop wise with constant stirring to precipitate Ni2+ as Ni -DMG once the precipitate is complete smell of NH3 should be distinct (this ensures the alkaline medium i.e. excess of ammonia). 4. Digest the precipitate on a boiling water bath for 20 - 30 minutes test the supernatant solution for complete precipitate with 1-2 drops of the DMG reagent as well as 1:1 NH3 solution 5. Filter the precipitate through a previously weighed Whatman filter paper No. 41 Standardization of EDTA solution : 1. Wash and clean burette I and burette II with water. 2. Rinse and fill the burette I with approx. EDTA solution (remove the air bubble) and place it at LHS. 3. Rinse and fill the burette II with 0.01M ZnSO4solution (remove the air bubble) and place it at RHS. 4. Take 10 ml of zinc sulphate solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. 5. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as X1 ml. 6. To the same solution in conical flask add 2 cm3of zinc sulphate solution from burette II and titrate with EDTA solution till clear blue solution obtained . Note down this reading as X 2ml. 7. Take one more reading in same way and record the reading as X 3 ml 8. From these three burette readings find out the exact molarity of EDTA solution. (C) Estimate the amount of Cu volumetrically:1. Transfer the precipitate in 250 ml of beaker. 2. Dissolve the precipitate by adding 10 ml conc. HCl in beaker and heat it to expel H 2S gas. 3. Then dilute the solution in a standard volumetric flask to 100 ml with distilled water and shake it well. 2. Rinse and fill the burette II with diluted Cu solution and place it at RHS. 3. Take 10 ml of diluted Cu solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. 4. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as Y1 ml. Or 4. Take 10 ml of diluted solution in conical flask. Add 5 – 7 cm3 of Liq. NH3 and 4 – 5 drops of 0.5% Fast sulphone black F indicator to it. 5. Titrate it against 0.01M EDTA solution from the burette till the colour changes from blue to dark green. Note down this burette reading. 6. Take two more readings. Find the constant burette reading as Y ml. Observation Weight of empty Whatman filter paper No. 41 = W1 g Weight of empty Whatman filter paper No. 41 + Ni-DMG = W2 g Weight of precipitate of Ni-DMG = W2-W1= X g Ni-DMG Practical Amount of Nickel in Ni DMG 288.71 g of Ni D.M.G. == contains 58.71 g of Nickel X g of Ni D.M.G == Xx 58.71 288.71 = A g of Nickel in given binary mixture Observation table For standardization of EDTA i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= 0.01MZnSO4 solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) 0.01MZnSO4 10ml 12 ml 14 ml Burette I (LHS) EDTA X1 = X2 = X3 = Calculations:Find out exact normality of EDTA EDTA = ZnSO4 M1 x X1 = 0.01 x 10 ∴M1 = 0.01 x 10 X1 M2 x X2 = 0.01 x 12 ∴M2 = 0.01 x 12 X2 M3 x X3 = 0.01 x 14 ∴M3 = 0.01 x 14 X3 Exact normality of EDTA= mean M4 = M1 M 2 M 3 3 For Estimation of Copper: i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= diluted Cu solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) diluted Cu solution Burette I (LHS) EDTA Determination of Cu: 1000 ml 1M EDTA. Y ml M4 M EDTA 10ml 10 ml 10 ml C.B.R Y1=____ ml Y2=____ ml Y3=____ ml Y= ____ ml = 63.55 g of Cu Yx M 4 x63.55 =? 1000 = B g of Cu. i.e. 10 ml diluted Cu solution contains = B g of Cu ∴100 ml diluted Cu solution contains = B x 10 = C g of Cu Results : (1) Amount of Nickel present in the given binary mixture solution =A= ________ g (2) Amount of Copper present in the given binary mixture solution =C= ________ g Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:________________________ Batch:____________ Aim : Separation of Cu-Zn mixture and estimate the amount of Zn gravimetric and Cu volumetric methods from the given binary mixture solution Theory: Binary mixture contain copper and zinc, in which copper is Group II radical and zinc is group IIIB radical. According to qualitative analysis technique separation of group II is done from the mixture.By adding dilute HCl and passing H2S gas copper get separated as CuS precipitate and filtrate is use for estimation of zinc . Requirements : Given binary mixture of Cu-Zn; Whatman filter paper No. 40, ethyl alcohol, , 0.01M ( approx.) EDTA solution, 0.01M ZnSO4, 25% diammonium hydrogen phosphate , NH4Cl,conc. HCl,1:1 NH3, EBT indicator, pH 10 buffer, Fast sulphone black F indicator Procedure : (B) Separtion of Cu-Zn mixture: 1. Dilute the given solution of mixture of Cu and Zn to 100 ml with distilled water in a standard measuring flask. Shake the flask well. Transfer it in a 250 ml beaker. 2. Add 20-25 ml dilute HCl ( two test tube) in it and warm the solution, then pass H2S gas through it till copper is completely precipitated. 3. Filter the black precipitate of CuS through ordinary filter paper .Use precipitate for estimation of copper and filtrate for estimation of zinc. (B) Estimate the amount of Zn gravimetric:1. Collect all the filtrate along with washing solution in 250ml beaker and add 1 ml conc. HCl, and boil it to expel H2S. 2. To this hot solution add 2 gm of NH4Cl and few drops of methyl red till solution becomes red. 3. Then add 1:1 NH3 drop wise till solution becomes yellow and heat it nearly to boiling. 4. Add 25% diammonium hydrogen phosphate till precipitation of Zn complete.( check by ppt. form in supernant liquid) 5. Filter the precipitate through a Whatman filter paper No. 40 and ignite it in previously weighed silica crucible. Standardization of EDTA solution : 1. Wash and clean burette I and burette II with water. 2. Rinse and fill the burette I with approx. EDTA solution (remove the air bubble) and place it at LHS. 3. Rinse and fill the burette II with 0.01M ZnSO4solution (remove the air bubble) and place it at RHS. 4. Take 10 ml of zinc sulphate solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. 5. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as X1 ml. 6. To the same solution in conical flask add 2 cm3of zinc sulphate solution from burette II and titrate with EDTA solution till clear blue solution obtained . Note down this reading as X 2ml. 7. Take one more reading in same way and record the reading as X 3 ml 8. From these three burette readings find out the exact molarity of EDTA solution. (C) 1. 2. 3. 4. 5. 6. 5. 6. 7. Estimate the amount of Cu volumetrically:Transfer the precipitate in 250 ml of beaker. Dissolve the precipitate by adding 10 ml conc. HCl in beaker and heat it to expel H 2S gas. Then dilute the solution in a standard volumetric flask to 100 ml with distilled water and shake it well. Rinse and fill the burette II with diluted Cu solution (remove the air bubble) and place it at RHS. Take 10 ml of diluted Cu solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as Y1 ml. Or Take 10 ml of diluted solution in conical flask. Add 5 – 7 cm3 of Liq. NH3 and 4 – 5 drops of 0.5% Fast sulphone black F indicator to it. Titrate it against 0.01M EDTA solution from the burette till the colour changes from blue to dark green. Note down this burette reading. Take two more readings. Find the constant burette reading as Y ml. Observation Weight of empty Crucible = W1 g Weight of empty Crucible + Zn2P2O7 ppt. = W2 g Weight of precipitate of Zn2P2O7 = W2-W1= X g Zn2P2O7 Practical Amount of Zinc in Zn2P2O7 ppt. 304.68 gm Zn2P2O7 == contains 130.74 gm of Zinc X g of Zn2P2O7 == Xx 130.74 304.68 = A g of Zinc in given binary mixture Observation table For standardization of EDTA i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= 0.01MZnSO4 solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) 0.01MZnSO4 10ml 12 ml 14 ml Burette I (LHS) EDTA X1 = X2 = X3 = Calculations:Find out exact normality of EDTA EDTA = ZnSO4 M1 x X1 = 0.01 x 10 ∴M1 = 0.01 x 10 X1 M2 x X2 = 0.01 x 12 ∴M2 = 0.01 x 12 X2 M3 x X3 = 0.01 x 14 ∴M3 = 0.01 x 14 X3 Exact normality of EDTA= mean M4 M1 M 2 M 3 3 = For Estimation of Copper: i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= diluted Cu solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) diluted Cu solution Burette I (LHS) EDTA Determination of Cu: 1000 ml 1M EDTA. Y ml M4 M EDTA 10ml 10 ml 10 ml C.B.R Y1=____ ml Y2=____ ml Y3=____ ml Y= ____ ml = 63.55 g of Cu Yx M 4 x63.55 =? 1000 = B g of Cu. i.e. 10 ml diluted Cu solution contains = B g of Cu ∴100 ml diluted Cu solution contains = B x 10 = C g of Cu Results : (1) Amount of Zinc present in the given binary mixture solution =A= ________ g (2) Amount of Copper present in the given binary mixture solution =C= ________ g Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_______________________ Batch:____________ Aim : Separation of Fe-Mg mixture and estimate the amount of Fe gravimetric and Mg volumetric methods from the given binary mixture solution Theory: Binary mixture contain iron and magnesium, in which iron is Group IIIA radical and magnesium is Group V radical. According to qualitative analysis technique separation of group IIIA is done from the mixture. By adding NH4Cl and NH4OH iron get separated as Fe(OH)3 precipitate and filtrate is use for estimation of magnesium. Requirements : Given binary mixture of Fe-Mg; Whatman filter paper No. 41, 0.01M ( approx.) EDTA solution, 0.01M ZnSO4, NH4Cl, 1:1 NH3, EBT indicator, pH 10 buffer Procedure : (C) Separation of Fe-Mg mixture: 1. Dilute the given solution of mixture of Fe and Mg to 100 ml with distilled water in a standard measuring flask. Shake the flask well. Transfer it in a 250 ml beaker. 2. Add 2gm NH4Cl stir well then add 1:1 NH3 drop wise till reddish brown precipitate is form. 3. Filter the precipitate of Fe (OH)3 through whatman filter paper No. 41 .Use precipitate for estimation of iron and filtrate for estimation of magnesium. (B) Estimate the amount of Fe gravimetric:1. Wash the precipitate with 10ml hot water. 2. Ignite the precipitate on metallic cone. 3. Transfer the residue along with paper in previously weighed silica crucible. 4. Heat it till reddish brown powder of Fe2O3 forms. Standardization of EDTA solution : 1. Wash and clean burette I and burette II with water. 2. Rinse and fill the burette I with approx. EDTA solution (remove the air bubble) and place it at LHS. 3. Rinse and fill the burette II with 0.01M ZnSO4solution (remove the air bubble) and place it at RHS. 4. Take 10 ml of zinc sulphate solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. 5. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as X1 ml. 6. To the same solution in conical flask add 2 ml of zinc sulphate solution from burette II and titrate with EDTA solution till clear blue solution obtained . Note down this reading as X 2ml. 7. Take one more reading in same way and record the reading as X 3 ml 8. From these three burette readings find out the exact molarity of EDTA solution. (C) Estimate the amount of Mg volumetrically:1. Boil the filtrate in 250 ml of beaker to make the volume below 100ml. 2. Then dilute the solution in a standard volumetric flask to 100 ml with distilled water and shake it well. 3. Rinse and fill the burette II with diluted Mg solution (remove the air bubble) and place it at RHS. 4. Take 10 ml of diluted Mg solution in a conical flask from burette II and add 10 ml of buffer solution of pH 10 and then add 2-3 drops of EBT indicator .Wine red colour develop to the solution. 5. Titrate the solution with the EDTA solution from burette I till wine red colour changes to blue colour. Note down this reading as Y1 ml. 6. Take two more readings. Find the constant burette reading as Y ml. Observation Weight of empty Crucible = W1 g Weight of empty Crucible + Fe2O3ppt. = W2 g Weight of precipitate of Fe2O3 = W2-W1= X g Fe2O3 Practical Amount of iron in Fe2O3 159.700 g of Fe2O3 == contains 111.700 g of iron X g of Fe2O3 == Xx 111.700 159.700 = A g of iron in given binary mixture Observation table For standardization of EDTA i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= 0.01MZnSO4 solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) 0.01MZnSO4 10ml 12 ml 14 ml Burette I (LHS) EDTA X1 = X2 = X3 = Calculations:Find out exact normality of EDTA EDTA = ZnSO4 M1 x X1 = 0.01 x 10 ∴M1 = 0.01 x 10 X1 M2 x X2 = 0.01 x 12 ∴M2 = 0.01 x 12 X2 M3 x X3 = 0.01 x 14 ∴M3 = 0.01 x 14 X3 Exact normality of EDTA= mean M4 = M1 M 2 M 3 3 For Estimation of Magnesium : i) Solution in burette I (LHS)= EDTA solution ii) Solution in burette II (RHS)= diluted Mg solution iii) Indicator = EBT iv) End point = wine red to blue Burette II (RHS) diluted Mg solution Burette I (LHS) EDTA Determination of Mg : 1000 ml 1M EDTA Y ml M4 M EDTA 10ml 10 ml 10 ml C.B.R Y1=____ ml Y2=____ ml Y3=____ ml Y= ____ ml = 24.01 g of Mg Yx M 4 x 24.01 =? 1000 = B g of Mg. i.e. 10 ml diluted Mg solution contains = B g of Mg ∴100 ml diluted Mg solution contains = B x 10 = C g of Mg Results : (1) Amount of iron present in the given binary mixture solution =A= ________ g (2) Amount of magnesium present in the given binary mixture solution =C= ________ g Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim : To prepare Tris (acetylacetonato) iron and purity determination. Theory :Tris (acetylacetonato) iron complex forms when Fe3+ reacts with acetyl acetone reagent in methanol which is a bidentate ligand with oxygen atom. By adding sodium acetate, acetate ions are made available which remove proton liberated by reagent. Transparent bright reddish brown crystals are formed which can be isolated by suction filtration. Requirements : FeCl36H2O, acetyl acetone reagent in methanol (7:3) CH3COONa, Whatman filter 41 etc. Procedure :Preparatrion of the complex: 1. Dissolve 2 g of ferric chloride hexahydrate in 10 – 15 cm3 of distilled water. 2. Add 10 – 12 cm3 of acetylacetone reagent in methanol gropwise with constant stirring. 3. After completion of addition of acetylacetone, stir it vigorously of 5 minutes. 4. Now, add in small portions, a solution of 3.6 g of sodium acetate dissolved in 10 cm3 of distilled water slowly with constant stirring. (or add 10 g of urea). 5. Transfer the beaker containing above mixture on a hot sand bath and heat it at 80oC with continuous stirring of 15 minutes. (If urea is used in above step then heat for 1 hour). 6. Cool to room temperature and then in an ice bath. 7. Filter off the reddish brown transperant crystals by suction and wash with 10 cm3 of cold distilled water. 8. Dry it by suction for 15 – 10 minutes and then by air. 9. Weigh the product to find out the yield (X g). Analysis of Tris (acetylacetonato) iron for the percentage of Iron: 1) Weigh 0.400 g of the complex on a watchglass and transfer quantitatively in a 250 cm3 beaker. 2) Add 5 cm3 of conc. HCl and heat it on a sand bath to decompose the complex and evaporate it to dryness. 3) Extract the residue with 25 cm3 of dilute HCl and dilute to 100 cm3 in a standard volumetric flask with distilled water. Shake the flask well for uniform mixing. 4) Fill burette no.1 with diluted solution of complex and burette no.2 with 0.025N K2Cr2O7 solution. 5) Take by burette no.1, 9 ml. of diluted complex solution in a100 ml. conical flask. 6) Add to it 2 ml. conc. HCl and boil it. It becomes yellow in colour. 7) To this hot solution add drop by drop 4% SnCl2 in excess to ensure complete reduction of ferric iron to ferrous iron. 8) Cool the solution in the flask thoroughly under tap water. 9) To this cooled solution add drop wise saturated solution of HgCl2 till a slight turbidity is just obtained. (Caution: Avoid excess of HgCl2 otherwise it will oxidize Fe2+ to Fe3+). 10) Titrate this solution with 0.025N K2Cr2O7 using K3 [Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X1 ml. 11) To the same solution, add 1 ml of diluted solution of complex from burette no.1. 12) Add 2-3 drops of conc. HCl and boil. 13) To this hot solution add carefully drop by drop SnCl2 solution( colorless) and then add one or two drops o f HgCl2 solution till a slight turbidity is just obtained. 14) Titrate it with 0.025N K2Cr2O7 solution using K3[Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X2 ml. 15) In a similar way take one more reading. Call this reading as X 3 ml. 16) From these readings calculate the percentage of iron in the complex. Observations and Calculations : (I)Yield of the complex: (A) Observed yield of the Complex (A) = _______ g. (B) Theoretical yield of the Complex = 270.30 g of FeCl36H2O = 353.17 g of [Fe(CH3COCH2COCH3)3] 353.17 x 2 ... 2 g of FeCl36H2O = 270.30 = 2.60 g of the Complex. (C ) Practical Yield 2.60 gm = 100% X gm = ? = 100x X 2.60 = ________% practical yield of complex Structure:- Reactions – 2FeCl3 + SnCl2 SnCl2 + 2HgCl2 (excess) K2Cr2O7 +14HCl + 6FeCl2 2FeCl2 + SnCl4 Hg2Cl2 + SnCl4 (turbidity)` 2KCl + 2CrCl3 + 6FeCl3 + 7H2O Amount of Fe in complex Observation table Given : K2Cr2O7 solution (0.025N) To find : Amount of Fe in the solution Burette 1 : Diluted complex solution Burette 2 : K2Cr2O7 solution Indicator : K3[Fe(CN)6] End point : No change in colour of indicator K3[Fe(CN)6] Burette-1 9 ml. 10 ml. Burette-2 X1 ml. X2 ml. 11 ml. X3 ml. Calculations : Estimation of Iron 1000 ml. 1N K2Cr2O7 = 55.84 g. of iron 1 ml. 0.025N K2Cr2O7 = 0.001396 g. of iron X1 ml. 0.025N K2Cr2O7 = X1x 0.001396 =A1 g. iron similary. X2 ml. 0.025N K2Cr2O7 = X2x 0.001396 =A2 g. iron andX3 ml. 0.025N K2Cr2O7 = X3x 0.001396 =A3 g. iron. Now, 9 ml. diluted solution contains A1 g. iron 100 xA1 100 ml. diluted solution contains = =B1 g. iron 9 Similary. 10 ml. diluted solution contains A2 g. iron 100 xA2 100 ml. diluted solution contains = =B2 g. iron 10 and 11 ml. diluted solution contains A3 g. iron 100 xA3 100 ml. diluted solution contains = =B3 g. iron 11 B1 + B2 + B3 3 = B g. of iron Purity of complex:353.17 g of [Fe(CH3COCH2COCH3)3] == 55.84 gm of Fe 0.400x 55.84 ... 0.400 gm of [Fe(CH3COCH2COCH3)3] = = 0.063 gm of Fe 353.17 ... 0.063 gm of Fe == 100% B g. of iron = 100x B = _________% purity of the complex 0.063 Results : (1) Yield of the Complex (X) = ___________ g. (2) Percent practical yield of complex = __________% (3)Percentage purity of the complex = _____% Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:________________________ Batch:____________ Aim : To prepare Potassium di aqua bis(oxalato) chromate (III) and purity determination. Theory :This experiment involved the preparation of chromium (III) complexes. The cis/trans isomeric pair provided an opportunity to look at isomerism in co-ordinate chemistry. Oxalic acid acted as the reducing agent in the syntheses. Optical activity can be generated only if it is possible to resolve a chiral compound into enantiomers. Enantiomers rotate the plane of plane polarised light in opposite directions by equal amounts. Resolution of chiral complexes is only achieved if the system is kinetically inert so that the process of ligand substitution is slow compared with the time taken to resolve the complex. For example the inert chiral complex [Co ( C 2O4)3]3- ( d6, low spin) can be resolved but the labile complex [Fe ( C2O4)3]3- ( d5, high spin) cannot. Resolution of complexes generally involves the presence of chelating ligands. Optically active cations can be resolved by reacting the mixture with an optically active anion to generate diastereoisomeric salt (different physical properties ). Requirements : Oxalic acid dehydrate, potassium oxalate monohydrate, solid K2Cr2O7 cthyl alchohol, Whatman filter paper No. 42, 2N H2SO4, 0.1 N (approx) Na2S2O3, 0.1 N (exact) K2Cr2O7 solution etc Procedure: (i) The preparation of potassium trans-diaquabis(oxalato)chromate(III) trihydrate. 3gm of oxalic acid dihydrate was dissolved dissolved in a 500cm3 beaker in the minimum amount of boiling water. To this a potassium dichromate solution was added made by dissolving 1g of potassium dichromate in the minimum amount of water. The potassium dichromate was added in small portions due to the violent reaction. When the reaction was completed the dark coloured solution was left to crystallise in a large beaker covered with a watch glass.The crude products must be washed with ethanol before drying because the alcohol itself is a dehydrating agent. It also gets rid of organic soluble impurities. Dry the crystals in air and weigh the dried product to find out the yield (x g). or (ii) Preparation of cis-K [ Cr (C2 O4)2 (H2 O)2 ]. 2H2O 12g of oxalic acid dihydrate was taken with 4g of potassium dichromate and they were ground into fine powders in the fume hood. Once mixing in the mortar was complete they were transferred to a 150cm3 beaker containing about 5 drops of water. The damp powder was then covered with a watch glass before a vigorous reaction occurred. Large volumes of carbon dioxide were evolved until only the dark coloured solution remained. 20cm3 of alcohol was added to the solution which was being continually warmed. After stirring the solution, a crystalline product eventually resulted. The product was filtered with a Buchner funnel, washed with alcohol and dried in the air. The yield was recorded (x g). Structure:- . Standardisation of 0.1 N (approx) solution 1. 2. 3. 4. 5. 6. 7. 8. Fill the burette-1 with 0.1 N (approx) Na2S2O3 solution and burette-2 with 0.1 N (exact) K2Cr2O7 solution. Take 10 ml K2Cr2O7 solution from burette-2 into conical flask/ stoppered bottle. Add to this solution 10 ml 10 % KI solution and 10 ml 2 N HCl. Stopper the bottle. Shake well. Keep the bottle in dark for 5 minutes to liberate iodine solution becomes brown. Titrate this solution with sodium thiosulphate solution slowly with constant shaking till solution becomes faint yellow. Now add to it 1 ml starch solution into it solution becomes blue. Continue addition of sodium thiosulphate drop by drop till the solution becomes colourless. Avoid excess addition of Na2S2O3. This will give rise error in burette reading. Note down this burette reading as X1 ml. To the same solution in bottle/conical flask add further 2 ml K2Cr2O7 solution from burette2 (total burette reading becomes 12 ml) and titrate with sodium thiosulphate solution from burette-1 till colourless solution is obtained. Note down this burette reading as X 2 ml. Repeat the procedure explained in point – vii once again and note third burette reading as X3 ml. Analysis of Potassium di aqua bis(oxalato) chromate (III) for the percentage of chromium 1. Weigh about 0.300 g of the complex on a watch glass and transfer it quantitatively in a 250 cm3 of beaker.Add 10 cm3 of 2N H2SO4 and heat it on a sand bath to decompose the complex. Extract the residue with 25 – 30 cm3 of distilled water and boil gently. 2. Cool the solution and transfer it quantitatively in a 100 cm3 standard measuring flask. Wash the beaker 2– 3 times with 5 cm3 of distilled water each time and transfer the washings in the same standard volumetric flask. Dilute the solution to 100 cm3 with distilled water. 3. Rinse the burette-1 and burette-2 with respective solution and fill the burette – 1 with 0.1 N Na2S2O3 and burette-2 with diluted Cr(III) solution. 4. Take 10 ml diluted Cr(III) solution from burette –2 in a conical flask/ stoppered bottle. Add to it 5. NH4OH drop by drop with constant shaking till slightly turbidity is obtained. Then add to it drop by drop with shaking acetic acid till turbidity dissolves giving clear solution. Add 1 ml of acid more to make it solution acidic. 6. Add to it 10 ml of 10 % KI solution. In this case I 2 is liberated. Shake the solution for 1-2 minutes and solution becomes brown. 7. Titrate this solution with sodium thiosulphate slowly with constant shaking till solution becomes faint yellow. 8. Now add 1 ml starch solution to it, solution becomes blue. 9. Continue addition of sodium thiosulphate drop by drop. From burette-1 blue colour disappears. Avoid excess addition of sodium thiosulphate. This will give rise error in burette reading. Note down this burette reading 10. Take two more readings. Find the constant burette reading as Y ml. Reactions : K2CrO7 + 7H2C2O4.2 H2O 2K[Cr(C2O4)2(H2O)2].2H2O + 6CO2 + 5H2O Observations and Calculations : (I) Yield of the Complex : (A) Observed yield of the Complex =X g. (B) Theoretical yield of the complex = 294 g of K2Cr2O7 = 678 g of the complex 2K[Cr(C2O4)2(H2O)2].2H2O 1 x 678 ... 1 g of K2Cr2O7 = 294 = 2.31 g of the complex 2K[Cr(C2O4)2(H2O)2].2H2O (C ) Percent Practical Yield 2.31 gm = 100% X gm a) = ? = 100 x X 2.31 = ________% practical yield of complex Standardisation of 0.1 N Na2S2O3 solution Observation and Observation Table – Burette 1 : 0.1 N Na2S2O3 solution Burette 2 : 0.1 N K2Cr2O7 Indicator : Starch. End Point : Blue to Colourless. Burette – 2 10 ml 12 ml 14 ml Burette – 1 X1 ml X2 ml X2 ml Calculation – Find out exact normality of sodium thiosulphate Na2S2O3 = N1 K2Cr2O7 = N 2 V2 X1 0.1 x 10 X1 N1x X1 = 0.1 x 10 ∴N1 = N2 x X2 = 0.1 x 12 ∴N2 = 0.1 x 12 X2 N3 x X3 = 0.1 x 14 ∴N3 = 0.1 x 14 X3 Exact normality of Na2S2O3 = mean N4 = N1 N 2 N 3 3 Determination Purity: a) Practically determined amount of Cr in (0.300 g) complex: 1000 ml 1 N Na2S2O3 = 17.07 g of Cr 17.07 xY xN 4 Y ml N4 Na2S2O3 =? g of Cr = 1000 = Z g of Cr (in 0.300 g of complex) b) Theoretical amount of Cr in (0.300 g) complex: K[Cr(C2O4)2(H2O)2].2H2O = Cr 339 g = 51.99 g Cr 0.300 g of K [Cr (C2O4)3].3H2O complex = ? = c) % purity of Cr in complex: 0.046 g of Cr Zg 0.300 x51.99 g of Cr 339 = 0.046 g of Cr = 100 % Purity Zx100 = = ______ % Purity 0.046 Results : (1) Yield of the Complex (X) = ___________ g. (2) Percent practical yield of complex = __________% (3) Percentage purity of the complex = _____% Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:________________________ Batch:____________ Aim: Synthesis of Chloropentamino Cobaltic chloride and determination of purity of the complex by estimation of cobalt. Principle:Cobalt forms various complexes. Most of them show that cobalt is in +3 oxidation state. It has coordination number 6. Cobalt is in centre surrounded by six coordinated ligands octahedrally. These ligands are H2O, NH3, Cl-, NO2-, ONO-, CO3-2 etc. The normal covalent compound contain divalent cobalt and in preparation of amino nitro cobalt (II) is easily oxidized to Co (III). H2O2 is used as oxidizing agent to get Cobaltic complexes in short time. Given:- CoCl2. 6H2O, NH4Cl, Liquor ammonia,conc. HCl,6 % H2O2, 0.05 N Na2S2O3,0.05N K2Cr2O7 10% KI Preparation of solutions: Preparation of Standard 0.05 N Dichromate Solutions: Weigh accurately 2.45 gm of K2Cr2O7 pure crystals 0n a watch glass, dissolve it in distilled water, and dilute it to 1000ml in volumetric flask. Preparation of Standard 0.05 N Sodium Thiosulphate Solutions: Weigh accurately 7.9 gm of N2S2O3 watch glass, dissolve it in distilled water, and dilute it to 1000 ml in volumetric flask. Procedure: (A) Synthesis of Chloropentamino Cobaltic chloride 1. Dissolve 5 gm of NH4C1 in 20 ml. of Liquor ammonia in beaker. 2. In another beaker (100 ml) take 2.5 g powdered Cobaltous chloride hexahydrate and add to it ammonium chloride solution in ammonia. 3. Then add from burette 8 ml of 20 volume H2O2 and stir well. 4. The remaining 8 ml of H2O2 is added drop wise (over period of 10 min) . 5. During the addition of H2O2 solution is stirred and the beaker is kept in cold water bath. Allow the reaction to complete for about 10—15 min. 6. Then neutralized by adding conc. HC1. Add more to make the whole solution acidic. 7. Test with Litmus Paper. Add 2 ml. of conc. HC1 in excess. Then heat the solution on wire gauge till boiling. Allow the solution to cool. Carmine red crystals are separated. Filter the precipitate on Buchner funnel at pump, wash with water to remove NH 4CI and then wash with ethanol & dry the crystals at room temperature under IR lamp. Weigh the yield. Reaction:[Co(H2O)6]Cl2 + NH4Cl + NH4OH 1000c [Co(NH3)5Cl]Cl2 + 6H2O. Stucture :- (B) Standardisation of 0.1 N Na2S2O3(approx) solution - 1. 2. Wash and clean burette I and burette II with water. Rinse and fill the burette I with approx. Na2S2O3 solution (remove the air bubble) and place it at LHS. Rinse and fill the burette II with 0.1N K2Cr2O7solution (remove the air bubble) and place it at RHS. Take 10 cm3 K2Cr2O7solution from burette II into conical flask/ stoppered bottle. Add to this solution 20 ml 10 % KI solution and 5 cm3 conc. HCl. Then add 20ml distilled water. Stopper the bottle. Shake well. Keep the bottle in dark for 5 minutes to liberate iodine solution becomes brown. Titrate this solution with Na2S2O3 solution slowly with constant shaking till solution becomes faint yellow. Now add to it 1cm3 starch solution into it solution becomes blue. Continue addition of Na2S2O3 drop by drop till the blue colour of solution disappears. Avoid excess addition of Na2S2O3. This will give rise error in burette reading. Note down this burette reading as X1cm3. To the same solution in bottle/conical flask add further 2 cm3 K2Cr2O7 solution from burette II (total burette reading becomes 12cm3) , solutions becomes blue colourthen titrate with Na2S2O3solution from burette I till blue colour of solution disappears. Note down this burette reading as X2cm3. Repeat the same procedure explained in point – vii to ix once again and note third burette reading as X3cm3. 3. 4. 5. 6. 7. 8. 9. 10. (C) Determination of purity of the complex by estimation of cobalt 1. Weigh accurately 0.1 gms. of Chloropentaminocobaltic chloride complex into 250 Conical Flask. 2. Add to it 15 ml. of 2 N solution of NaOH and heat it on wire gauge till ammonia is liberated completely and black Cobaltic oxide is obtained. 3. Filter it through Whattman filter paper & wash it with hot water to remove Cl- ion completely. Then take paper along with residue into the conical flask. 4. Add to it 8 ml. of Conc. HC1, 0.33 gm of KI (potassium Iodide) and add 15 ml distilled water. Press the cork and shake it well and keep it in the dark place for 10 min. When all black particles of Cobaltic oxide have dissolved. Titrate it with Sodium thiosulphate solution (0.05 N) using starch solution as an indicator. The end point is Blue to faint pink coloration.Note that reading as Y ml Calculations: Standardisation of 0.1 N Na2S2O3solution Observation and Observation Table – Burette 1 Burette 2 Indicator End Point : : : : 0.1N( approx.) Na2S2O3 solution 0.1 N K2Cr2O7 Starch. Blue colour disappears. Burette – 2 10 cm3 12cm3 14cm3 Burette – 1 X1=___ cm3 X2=___ cm3 X3=___ cm3 Calculation – Find out exact normality of sodium thiosulphate Na2S2O3 = K2Cr2O7 ∴ N1 x X1 = 0.1 x 10 N1 = N2 x X2 = 0.1 x 12 ∴ N2 = N3 x X3 = 0.1 x 14 ∴ N3 = 0.1 x 14 X3 Exact normality of Na2S2O3= mean N4 = 0.1 x 10 X1 0.1 x 12 X2 N1 N 2 N 3 3 Observation:Weight of product = W gm = ____ gm. Calculations: 1) Theoretical yield: CoCl2.6H2O 237.95 g of CoCl2.6H2O 2.5 g of CoCl2.6H2O 2) % Practical yield: = = = [Co (NH3)5.Cl] Cl2 250.45 g of [Co (NH3)5.Cl] Cl2 2.63 g of [Co (NH3)5.Cl] Cl2 2.63 g = 100 % W g =? = W x 100 2.63 = -------- % 6) Purity of cobalt from the complex: a) Practical yield of Co in complex: 1000 ml 1 N Na2S2O3 Y ml N4 Na2S2O3 = 58.94 g of Co = 58.94 x Y x N4 / 1000 = Z g of Co b) Theoretical yield of Co in complex: 250.45 g [Co (NH3)5Cl] Cl2 complex ≡ 58.94 g of Co 100 g [Co (NH3)5Cl] Cl2 of complex = 58.94 x 100 g of Co 250.45 = 23.53 g of Co Therefore, 0.1 g [Co (NH3)5Cl] Cl2 of complex = ? = 0.1x 23.53 100 = 0.023 g c) % practical yield of Co in complex: 0.023 g of Co = 100 % Purity Therefore, Therefore, Z g of Co = Z x 100 0.023 = ______ % Purity Results : (1) Yield of the Complex (X) = ___________ g. (2) Percent practical yield of complex = __________% (3) Percentage purity of the complex = _____% Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:__________________ Batch:______________________________ Aim : Estimation of iron from given drug sample Chemicals : Conc. HCl, Conc. HNO3,Perchloric acid (60%), K3[Fe(CN)6] indicator, K2Cr2O7 solution (0.025N) , SnCl2 solution (4%), HgCl2 solution (saturated). Apparatus : Stem-cut funnel ,100 ml volumetric flask, beaker, burettes, etc Procedure: 1) Weigh accurately about one gram of iron tablet. Call it as W g. and transfer it into a crucible 2) Cover the crucible with led with slightly opening. Heat first at low flame till all organic matter is evolved out. 3) Then heat on strong flame, till complete ash is formed. 4) Tranfer the ash in conical flask and add to it about 15 to 20 ml conc. HCl. 5) Place a stem-cut funnel on the mouth of the conical flask which acts as an air-condenser. 6) Heat the flask in a fuming cupboard on a low flame till the ash dissolves. If the ash does not dissolve add 1 or 2 ml conc. HCl, 5ml water and heat it again on a low flame. 7) Cool the flask and add 2 to 3 ml. conc. HNO3 and heat it again for 2 to 3 minutes. 8) Cool the flask and add 5 ml. 60% perchloric acid to it. Heat it again till white dense fumes start evolving. ( perchloric acid is a powerful oxidizing agent. It is used here to oxidize ferrous iron to ferric iron). 9) Cool the flask, rinse the funnel and its stem with little water into the conical flask. 10) Add to it one test tube of distilled water. 11) Filter the above solution through Whatman No.41 filter paper to remove impurity if any. Collect the filtrate in a beaker. Dilute it to exactly 100 ml in a volumetric flask. Use this Solution for the estimation of iron. Preparation of 0.01 N K2Cr2O7 1000ml 1N === 49gm K2Cr2O7 1000ml 0.025N === 1.225 gm K2Cr2O7 Part II : Volumetric estimation of iron : Fill burette no.1 with diluted solution of iron and burette no.2 with 0.025N K2Cr2O7 solution. 11. Take by burette no.1, 9 ml. of diluted iron solution in a100 ml. conical flask. 12. Add to it 2 ml. conc. HCl and boil it. It becomes yellow in colour. 13. To this hot solution add drop by drop 4% SnCl2 in excess to ensure complete reduction of ferric iron to ferrous iron. 14. Cool the solution in the flask thoroughly under tap water. 15. To this cooled solution add drop wise saturated solution of HgCl2 till a slight turbidity is just obtained. (Caution: Avoid excess of HgCl2 otherwise it will oxidize Fe2+ to Fe3+). 16. Titrate this solution with 0.025N K2Cr2O7 using K3 [Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X1 ml. 17. To the same solution, add 1 ml of iron solution from burette no.1. 18. Add 2-3 drops of conc. HCl and boil. 19. To this hot solution add carefully drop by drop SnCl2 solution .Add one or two drops o f HgCl2 solution till a slight turbidity is just obtained. 20. Titrate it with 0.025N K2Cr2O7 solution using K3[Fe(CN)6] as an external indicator. The end point of the titration is noted when there is no change in colour of the indicator. Call this reading as X2 ml. 21. In a similar way take one more reading. Call this reading as X 3 ml. 22. From these readings calculate the percentage of iron in the tablet. Given : K2Cr2O7 solution (0.025N) To find : Amount of Fe in the solution Burette 1 : Diluted Fe solution Burette 2 : K2Cr2O7 solution Indicator : K3[Fe(CN)6] End point : No change in colour of indicator K3[Fe(CN)6] Reactions – 2FeCl3 + SnCl2 SnCl2 + 2HgCl2 (excess) K2Cr2O7 + 14HCl + 6FeCl2 Burette-1 Burette-2 9 ml. X1 ml. 2FeCl2 + SnCl4 Hg2Cl2 + SnCl4 (turbidity)` 2KCl + 2CrCl3 + 6FeCl3 + 7H2O 10 ml. X2 ml. 11 ml. X3 ml. Calculations : Estimation of Iron 1000 ml. 1N K2Cr2O7 = 55.84 g. of iron 1 ml. 0.025N K2Cr2O7 = 0.001396 g. of iron X1 ml. 0.025N K2Cr2O7 = X1 x 0.001396 = A1 g. iron similary. X2 ml. 0.025N K2Cr2O7 = X2 x 0.001396 = A2 g. iron and X3 ml. 0.025N K2Cr2O7 = X3 x 0.001396 = A3 g. iron. 9 ml. diluted solution contains A1 g. iron 100 xA1 100 ml. diluted solution contains = =B1 g. iron 9 Similary. 10 ml. diluted solution contains A2 g. iron 100 xA2 100 ml. diluted solution contains = =B2 g. iron 10 and 11 ml. diluted solution contains A3 g. iron 100 xA3 100 ml. diluted solution contains = =B3 g. iron 11 B1 + B2 + B3 = B g. of iron 3 Now, W g of tablet = ___________ B g. of iron 100 xB 100 g of tablet = = ____________ % iron W Result :- 1] Weight of given tablet = W=____________ gms 2] Iron content in tablet =B= ____________ gms 3] Percent of Iron = _____________ % Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim : Determination of heat of dissolution and hence lattice energy of the salt NaCl, KCl and CaCl2. Theory : Heat of dissolution of a salt is defined as the amount of heat evolved or absorbed when one gram mole of the salt is dissolved in 1000 ml. of the solution, at a given temperature. It is an experimental quantity. Lattice energy, heat of dissolution and heat of hydration of a salt are related by Born-Haber equation of follows : ∆Uo = ∆Hoh - ∆Hos + 2RT x 10-3 ……..(1) Where ∆Uo = Lattice energy of the salt o ∆H s = Standard heat of dissolution of the salt ∆Hoh = Standard heat of hydration of the salt ∆Hoh = ∆Hos (c) + ∆Hos (a) Where ∆Hos (c) = Std. heat of hydration of cation ∆Hos (a) = Std. heat of hydration of anion If the value of Hoh is given for a salt, if temperature T is known and if Hos is experimentally found out Uo can be found out. Equation I holds true for a 1:1 electrolyte like NaCl and KCl. This equation changes for a 1:2 electrolyte like CaCl2 as ∆Uo = ∆Hoh - ∆Hos + 3RT x 10-3 Chemicals : Solids - NaCl, KCl, CaCl2, distilled water. Apparatus - Dewar flasks, Thermometer, 100 ml. measuring cylinder, 250 ml. beakers. Procedure : The experiment consists of two parts. a) Determination of water equivalent of the Dewar flask 1) Take 100 ml. of distilled water in a Dewar (Thermos) flask. Note its temperature after 5 minutes. Call it toc. 2) Take about 110 ml. distilled water in a 250 ml. beaker and heat it to about 45 oC. 3) Take 100 ml. of this heated water in a measuring cylinder. Record its temperature. Call it toh (It will be around 40oC). 4) After recording the temperature of heated 100 ml. water, pour this hot water (100 ml.) from measuring cylinder to Dewar flask quickly. 5) Stir the mixture in the Dewar flask with the help of thermometer 4 to 5 times and record the steady temperature of this water. The temperature will become steady within 30 to 60 seconds. Call this temperature tm. 6) Find the water equivalent W of the Dewar flask. 7) Repeat this procedure to find the water equivalent W of the Dewar flask once again. b) Determination of heat of dissolution of the salt 1) Take 100 ml. distilled water in a Dewar (Thermos) flask. Note its temperature after 5 minutes. Call it t oi 2) Weigh exactly 4 grams solid NaCl and put it in the 100 ml. water taken in the Dewar flask. 3) Gently stir with the thermometer till the salt dissolves. 4) When no solid particles are seen at the bottom of the Thermos flask, immediately record the temperature of the solution. Call it t of 5) Repeat this experiment for 4 grams NaCl once again. 6) Calculate the heat of dissolution of NaCl using the above formula. 7) Perform the same experiment twice using 4 grams of KCl. 8) Perform the same experiment twice using 4 grams of CaCl2 9) Calculate the heat dissolution of KCl and CaCl2 using proper formulae. Observation Table : NO Temp. of Cold water toc Temp. of hot water toh Temp. of the mixture 200 ml. tom 1 2 Determination of heat of dissolution of the Salt No. Salt Initial Final Temp of Temp. of water water toi tof 1 NaCl 2 NaCl 3 KCl 4 KCl 5 CaCl2 6 CaCl2 Water equivalent W o ∆H s Average ∆Hos Average W Expected value ∆Hos Calculations : Determination of water equivalent of Dewar flask : Heat lost by hot water = Heat gained by cold water + Heat gained by Dewar flask. 100(th – tm) = 100(tm – tc) + W (tm – tc) 100(th – tm) - 100(tm – tc) = W (tm – tc) 100[th + tc – 2tm ] --------------------- = W grams (tm – tc) Using this formula, find the water equivalent of the Dewar flask twice. The two values should be near about the same. Take the mean of the two values of W. 1) Determination of heat of dissolution of NaCl dq = (100 + W) (tf – ti) = (100 + W) (∆t ) where dq is the heat evolved or absorbed W is the average water equivalent When t is negative, heat change is positive and the process is endothermic. When t is positive, heat change is negative and the process is exothermic For 4g NaCl = heat change is dq dq x 58.5 For 58.5 g NaCl = heat change is --------------- = ∆Hos 4 (Mole wt. of NaCl. = 58.5) ∆Ho (in cal) x 4.184 = Hos Joules/mole Convert it to kJ/mole by dividing by 1000. 2) Determination of heat of dissolution of KCl Molecular Weight of KCl = 74.5 Calculate the heat of dissolution of KCl in a manner similar to that of NaCl. 3) Determination of heat of dissolution of CaCl2 Molecular Weight of CaCl2 = 111 Calculate the heat of dissolution of CaCl2 in a manner similar to that of NaCl. Use the equation ∆Uo = ∆Hho - ∆Hos + 2RT x 10-3 for NaCl and KCl and ∆Uo = ∆Hoh - ∆Hos + 3RT x 10-3 for CaCl2 Given - ∆Hoh for NaCl = - 790 KJ/mole KCl = -700 KJ/mole CaCl2 = - 2340 KJ/mole R = 8.314 J/oC/mole T = Room Temp oC + 273 o ∆H s = Experimentally found out forNaCl, KCl and CaCl2. Calculate the value of Uo using the above values and equation. The expected value for ∆Uo for NaCl = -777.8 kJ/mole for KCl = -708.8 kJ/mole for CaCl2 = - 2255.0 kJ/mole Result Table : Salt NaCl KCl CaCl2 ∆Hos Experimental ∆Uo Experimental Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim:- Determination of the Rf value of Pb,Cu,Cd ions by using paper chromatographic technique Theory :- The separation of inorganic compounds is base on their different solubilities in organic solvent. In some cases solubility is due to formation of soluble complex between the inorganic compound and organic solvent, whereas in other cases complex forming reagents deliberately added. Apparatus and Solutions:- Whatman No.1 Paper Strip (25x7cm) chromatographic jar for ascending chromatography (30cm height x 10cm in diameter) ; 1% Solutions of CuCl2, Pb(NO3)2 and CdCl2 with few drops of dilute HNO3 (to prevent hydrolysis); the mobile phase may be ethanol (90cm3) + 5N HCl (10cm3), or 1-butanol saturated with 3N HCl, or ethanol (45cm3) + 2-propanol (45cm3) + 5N HCl (10cm3). Visualising reagent may be colourless (NH4)2S Solution, prepared by passing H2S in Dilute NH4OH for sufficient time, or a 0.05% dithiozone solution in CHCl3 and pass H2S,025M K2CrO4 and expose to ammonia ,and melting point capillaries. Procedure:1. Obtain a filter paper strip and draw an origin line by pencil about 3cm away from one end parallel to the breadth. 2. Put the x marks and least 2cm away from each other on this line. 3. At various points, solutions of cations (to be separated) are applied 5-6 times by capillaries and dried The paper strip (after drying) is then placed in the jar (ascending mode). 4. It is then lowered slowly into the developing solvent(all the spots remain above the solvent boundary) so that the lower 1.5cm edge of the strip dips in it. 5. The jar is then covered properly and tightly. 6. When the solvent front has travelled a reasonable distance (say 18-20cm), the strip is removed from the jar, the solvent front is marked with a pencil. 7. It is then dried. After Drying When the spots are sprayed with 0.05% dithiozone in CHCl3, the various spots show colours as: Cu-purple brown; Pb-rose pink; Bi and Cd-purple and Hg(II) – pink. When the spots are sprayed with colourless (NH4)2S solution all sports appear black except that due to Cd(II) which is yellow. After Spraying the Spots, calculate the Rf values for different cations using different mobile phases comes out to be different. Solvent (Ethanol + Distilled water in 7:3): Cd (II) > Cu (II) > Pb (II) Solvent (n- Butanol + glacial Acetic acid in 6:1) : Cd (II) > Pb (II) > Cu (II) Solvent (Acetone + Distilled water + conc. HCl in 17:1:2): Cd (II) > Cu (II) > Pb (II) Calculations: Determination of Rf values by using formula :Rf = distance travelled by cation/distance travelled by solvent RESULT:Cation Colour develop Rf value Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim:- Determination of the Rf value of Fe,Al,Cr ions by using paper chromatographic technique Theory :- The separation of inorganic compounds is base on their different solubilities in organic solvent. In some cases solubility is due to formation of soluble complex between the inorganic compound and organic solvent, whereas in other cases complex forming reagents deliberately added. Apparatus and Solutions:- The Whatmann No.1 paper strip and chromatographic jar as before. The mobile phase is ethanol (45cm3) + 2-propanol (45cm3) + 5N HCl (10cm3). Aqueous 1% solution of ferric chloride, aluminium chloride and chromium chloride are prepared. A few drops of HCl are added to prevent hydrolysis. The locating reagent is 1%alcoholic alizarin solution and 0.05% solution of benzidine in acetic acid and melting point capillaries. Procedure:Apply the sample and develop the chromatogram of group III cations as in previous exercises. After the solvent has evaporated and the strip has perfectly dried, it is cut lengthwise into two portions. One portion is sprayed with a saturated alcoholic solution of alizarin, made alkaline by exposure to ammonia vapours and then warmed: Al(III) appears as a red band well separated from a purple band due to Fe(III). The other portion of the strip is sprayed first with 5% aqueous sodium peroxide and then with 0.05% solution of benzidine in 10% acetic acid; Cr is located as a bright blue band just behind the Al. The sequence of Rf values in this separation is as; Fe(III)> Al(III)>Cr(III) Calculations: Determination of Rf values by using formula :Rf = distance travelled by cation/distance travelled by solvent RESULT:Cation Colour develop Rf value Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim: To titrate the given mixture of CO32- and HCO3 – ions against a strong acid (HCl) using pH meter and to determine the strength of it. Theory: When the solution containing CO32- and HCO3- ions are titrated against strong acid like HCI, initially CO32- ions react with HCI and form HCO3- ions and when half of the CO32- ions are neutralised , there will be sudden and sharp decrease in pH. Then all continuation of titration other half CO32- ions form HCO3 - ions and finally all the HCO3- ions are neutralised. Again sudden fall in pH will be observed form the graph of pH against volume of HCI added, volume of HCI required to neutralise CO32- and HCO3- ions is found out, hence strength of it in a mixture. Requirements : 0.1 N HCl, mixture of CO32- and HCO3- ions, distilled water, buffer solutions of 0.05N K.H.P pH 4.00, volumetric flask,100ml beaker, burette,10ml pipette. Reactions: (i) Na2CO3 + HCl NaHCO3 + NaCl (ii) NaHCO3 + HCl NaCl + H2O + CO2 Procedure 1. Standardized the pH meter by 0.05N K.H.P at pH 4.00. and pH 7.00 by water. 2. Dilute the given solution of mixture of CO32- and HCO3- ions to 100 ml with distilled water in a standard measuring flask. Shake the flask well. 3. Pipette out 10 ml of the diluted solution in a 100 ml beaker and dip the electrode in it [ Add distilled water if required to dip the electrodes] 4. Read pH value from the scale and record it in a tabular form. 5. Now, add 0.5 ml of 0.1 N HCl solution from the burette. Stir the solution and note down the pH value 6. Repeat step No 5 until there is a sudden fall in pH values. This sudden fall in pH is due to the half neutralization of CO32- ions 7. Continue titration in same way till the pH values show a sudden fall a second time which is due to the complete neutralization of the remaining CO32- ions and finally all the HCO3 – ions Take 6-8 reading more. 8. Plot a graph of pH against volume of HCl added. Observation 1. Volume of diluted mixture of CO32- and HCO3 – titrated = 10 ml 2. Normality of HCI used = 0.1 N Observation Table Observation Nos. Volume of 0.1N HCl added in ml pH 1 0.0 2 0.5 3 1.0 | | so on so on Graph : Plot the graph against the volume of HCl added. Calculations: 1. From graph, volume of HCl required to neutralise CO32- ions completely = 2Vx1 Acid ------ Alkali i.e. HCl - Na2CO3 N1V1 = N2V2 0.1 x 2Vx1 = N2 x 10 . .. 2. 0.1 x 2Vx1 N2 = ------------ ------ = A N 10 g/litre of Na2CO3 = N2 x equivalent weight = A x 53 = x g. . . . 100 ml of the diluted solution will contain 10x g of Na2CO3 53 g of Na2CO3 = 30 g of CO32- ions 30 x 10x ... 10x g of Na2CO3 will contain ------------ = _____g of CO3253 From graph, volume of HCl required to neutralize both CO32- and HCO3 - ions completely = Vx2 ... Volume of HCl required to neutralise only HCO3= Vx2 – 2Vx1 = z ml. Acid ------ Alkali i.e HCl – NaHCO3 N1V1 = N3V3 0.1 x z = N3 x 10 0.1 x z . .. N 3 = ---------------=BN 10 ... g/litre of NaHCO3 = N3 x equivalent weight = B x 84 = y g ... 100 ml of the diluted solution will contain 10y g of NaHCO 3 84 g of NaHCO3 = 61 gms of HCO3- ions 61 x 10 y ... 10 y g of NaHCO3 will contain. -----------= g of HCO3- ions 84 Results: 1 ] 10 ml of the diluted mixture solution requires = ……. ml 0.1N HCl solution for neutralization of Na2CO3 solution. 2] 10 ml of the diluted mixture solution requires = ……. ml of 0.1N HCl solution for neutralization of NaHCO3 solution. 3] Amount of CO32- ions present in a given solution = ……. g. 4] Amount of HCO3- ions present in a given solution = ……. g. Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim: Flame photometric determination of Ca by calibration curve and standard addition method. Theory: In a calibration curve method the flame is aspirated with known concentration of the element to be determined and respective readings are recorded. A calibration curve is then constructed by plotting signal intensity for each of these standard solutions against the respective concentration. Then the sample having unknown concentration is aspirated and signal intensity is recorded. On the basis of signal intensity concentration of element in simple can be obtained. If necessary, the test solution may suitable be diluted to get the readings in the range of 0.1 to 0.4. The calibration curve should be checked periodically by making measurements with the standard solutions. Requirments 100 ppm calcium ion solution Procedure II. 1. Take five different volumetric flasks marked as 1, 2, 3, 4 and 5 2. Add 5, 10, 15, 20 and 25 ml of stock solution of Ca to these flasks respectively. 3. Dilute the solutions with distilled water to obtain solutions of different concentrations (as given in the observation table.) 4. Switch on the instrument and wait for at least ten minutes. 5. Adjust the sensitivity control to the minimum value. 6. Turn the gas supply fully on and light the gas at the burner. 7. Adjust the air supply from the compressor until the pressure indicated on the pressure gauge attains a value of 10 lbs/sq. inch (0.7 kgs/6m2). 8. Charge the sample beaker with distilled water and place it in position so that the atomizer intake tube dips into the water. The liquid is thus drawn up the inlet tube by the stream of air and is atomized to a fine mist. 9. Regulate the gas supply so that the blue cone of the flame forms separate cones to each burner hole. 10. Place the appropriate filter in calcium=662 11. Spray a standard solution containing the ions to be determined and adjust the sensitivity controls to get a deflection of 100 divisions (full scale) on the meter for highest concentration.(orange colour to flame) 12. Spray distilled water and adjust the galvanometer to read zero by means of zero control. 13. Spray the standard solution again and read just the sensitivity control for full-scale deflection of the galvanometer. 14. Check the zero by spraying with distilled water again. 15. Spray the various solutions of known concentrations prepared in step No.3 and note the galvanometer readings at each concentration. 16. Plot a graph of galvanometer readings against concentrations of solutions to form the calibration curve. Reading for sample solution : 17. Dilute the given sample solution of unknown concentration to 100 ml in a standard measuring flask numbered as 6 with distilled water. 18. Spray the sample solution of unknown concentration in the flame. Note the galvanometer deflection (or reading). 19. Evaluate the concentration from the calibration curve. Observations : Flask No. 1 2 3 4 5 6 Volume of stock solution in ml. 05.0 10.0 15.0 20.0 25.0 (sample) Total volume of Concentration solution in ml of solution in ppm 100 100 100 100 100 100 Reading 05 10 15 20 25 X Graph : Plot a calibration curve for Ca with the help of the readings obtained for various solutions of known concentrations Results 1. Concentration of Ca+2 ions in the given sample solution 2. Amount of Ca+2 ions in the given sample solution = …….ppm. = …….. g. Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim: To estimate the percentage of Manganese present in a sample of mild steel calorimetrically. Principle: The given steel sample is dissolved in nitric acid. Manganese (II) is oxidized to permanganic acid by heating with lead peroxide and conc. Nitric acid. The intensity of the permanganic acid is measured in the colorimeter and the amount of manganese is estimated. Requirements : Steel sample,Nitric acid (HNO3), Syrupy phosphoric acid(H3PO4),Lead peroxide , 100 ml standard flask, Pipette,Measuring cylinder, Funnel, Colorimeter Procedure: 1. Dissolution of the steel sample: Weight out about 0.5 grams (500 mg) of the given steel sample into a clean 250 ml conical flask and add about 30 ml of 1:3 nitric acid heat to boiling, till the sample goes into solution. Boil gently for about 10 to 15 minutes, to oxidize any carbon present, cool and transfer into a clean 100 ml standard flask. Wash the conical flask twice with about 10 ml portions of distilled water and transfer into the standard flask. Make up the solution upto the mark with distilled water and shake well for uniform concentration. 2. Development of Colour: Transfer 20 ml of the prepared solution into a clean conical flask add about 10ml of syrupy phosphoric acid and about 20 ml of 1:3 nitric acid and heat the solution to boiling. Add two spatulas of lead peroxide and continue the boiling for about 5 minutes. Take out the flask from the stand and allow it to cool and settle. Decant the supernatant liquid into a 100 ml standard flask, wash the precipitate twice with little distilled water and collect the supernatant liquid and the washings into the standard flask and make up the solution with distilled water and shake well uniform concentration. Transfer about 10 ml of the solution into a clean centrifuge tube and centrifuge the solution to separate out any lead peroxide particles. Measure the optical density in the colorimeter and calculate the amount of Manganese from the standard calibration curve. Weight of the sample: W1 = Weight of bottle + sample = ____________ gms W2 = Weight of bottle = ____________ gms Weight of sample = (W1-W2) = ____________ gms. Standard value for calibration curve Amount of Manganese (in mg) O. D 0.1 0.035 0.2 0.065 0.3 0.100 0.4 0.130 0.5 0.155 0.6 0.190 0.7 0.220 0.8 0.245 0.9 0.260 = ____________ mgs X = Optical density of the solution = Y = Corresponding value of Mn from the curve = Calculations: Percentage of Manganese present in the steel sample = Y x 100 x 100 20 x (W1-W2) mg Result: Percentage of Manganese present in the mild steel sample is = Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim:- To estimate the amount of copper present in a given solution by iodometric method using potentiometer. Theory : In this method, a cell is set up which consists of calomel as reference and an oxidation reduction electrode as the indicator electrode. The indicator electrode used is sensitive to iodide ion concentration which is madeup of excess of I2.I2 gets reduced during the course of titration. The change in electrode potential is due to change in activities of I o and I- ion in the solution; which change the electrode potential of electrode. Amount of I2 liberated depends upon the concentration of copper ion present in the solution. The reaction involved is : 2Io + 2S2O32- ----- 2I- + S4O62Requirements : Potentiometer, platinum electrode, calomel electrode, KCl salt bridge, burette, pipette, distilled water Liq. NH3. glacial acetic acid, 10% KI, 0.05N Na2S2O3 solution, unknown concentration copper solution etc. Procedure 1. Dilute the given solution of CuSO4.5H2O to 100 ml with distilled water in standard measuring flask, Shake well. 2. Pipette out 10 ml of diluted solution in a 100 ml beaker. Add liquor ammonia solution drop wise to it till the dark blue colored cuprammonium complex forms add 2 – 3 drops more. 3. Decompose the cuprammonium complex with glacial acetic acid and add 1 or 2 drips more. 4. Add 20 ml of 10% KI solution to it and stir well. Insert a platinum wire [add distilled water, if necessary, until the platinum electrode is fully immersed] and connect it to the positive terminal of potentiometer. 5. Take saturated KCl solution in another beaker after cleaning and rinsing it. 6. Place a saturated calomel electrode in KCl solution and connect it to the negative terminal of the potentiometer. 7. Put a KCl salt bridge connecting the two beakers. This cell is represented as follows: (Negative terminal) Saturated calomel electrode || I- | Pt, I2+ (positive terminal) 8. standardize the potentiometer and measure e.m.f. of the cell 9. Add 1 ml of 0.05 N Na2 S2O3 solution from the burette to the solution in the heater. Stir it well. 10. When dark yellow solution starts changing its colour to whitish yellow colour, add 0.2 ml of Na2 S2O3 solution (change in colour indicates end point is nearer. Stir the solution thoroughly, after each addition) 11. continue to add 0.2 ml of Na2 S2O3 solution, till the decrease in e.m.f. between two successive readings is very small or nearly constant Record the reading 12. Plot a graph of e.m f. against volume of Na2 S2O3 solution added and E/V against the volume of Na2S2O3 solution added, and determine the volume Vx required for the equivalence point. Observations and calculations : Obs.No. Volume of e.m. of cell e Difference Difference ∆E Na2S2O3 in volts in e.m.f. ∆E in volume ---- mill solution in milli volts ∆V in ml ∆V added V ml Volt/ml 1 Vo Eo 2 V1 E1 E1 – Eo V1 – Vo 3 V2 E2 E2 – E1 V2 – V1 4 V3 E3 E3 – E2 V3 – V2 . . . Graphs: 1. e.m.f. against volume of Na2S2O3 solution added. E 2. against volume of Na2S2O3 solution added. V Reactions (i) 2CuSO4 + 4KI ------ Cu2I2 + 2K2SO4 + I2 (ii) I2 + 2Na2S2O3 ------ Na2S4O6 + 2NaI Calculations Volume of NaS2O3 solution required for equivalence point: (i) from graph of e.m.f. against volume of titrant added = Vx1 ml = ……… ml. (ii) from graph of ∆E against volume of Na2S2O3 solution added. ∆V = Vx2 ml = ……… ml. Mean = (Vx) = V x1 V x 2 = ……… ml. 2 From reactions, 2Na2S2O3 = 2I = 2Cu = 2CuSO4.5H2O. ... Na2S2O3 = I = Cu = CuSO4.5H2O. 1000 ml of 1N Na2S2O3 = 63.54 g of copper. 63.54X 0.05X 10Vx 10 Vx ml of 0.05N Na2S2O3 = 1000 = ………. g of copper. Results 1. Volume of Na2S2O3 required for the titration of 10 ml of diluted copper solution Vx =... ml. 2. Amount of copper present in the given solution = ……… g. Department of Chemistry Pratap College, Amalner Experiment No.: ________ Date:____________ Name of Student:_____________________________ Batch:____________ Aim: To determine the amount of boric acid present in the given solution by titrating against weak base conductometrically. Theory: It is a titration of weak acid against a weak base. Boric acid acts as a weak monobasic acid and is difficult to titrate against a standard alkali solution. However, it can be titrated conductometrically. The initial conductance is very small but it increases as the neutralization proceeds due to formation of salt, after neutralization, excess of NH4+ ions shows common ion effect and hence conductance becomes almost constant. The point of intersections in the graph gives the end point of the titration. Requirements: Conductometer and cell, burette, beaker, pipette, conductivity water, 0.1N NH4OH solution, boric acid solution etc. Procedure: (A) Standardization of NH4OH Solution: 1) Pipette out 10 ml of 0.1N(approximate) or supplied NH4OH solution in a conical flask and add 2-3 drops of methyl orange to it. 2) Titrate it against 0.1N HCl solution from the burette till the colour changes from yellow to reddish orange.(say X ml) 3) By using N1V1=N2V2 formula, calculate exact normality of NH4OH solution. (B) Estimation of Boric acid: 1) Dilute the given solution of boric acid to 100ml by using conductivity water in a standard measuring flask, Shake the solution well. 2) Pipette out 10 ml of the diluted solution in a 100 ml beaker. 3) Place the conductivity cell in the above beaker and add sufficient quantity of conductivity water to immerse the black plates of electrode completely. 4) Insert a glass rod to stir the solution well, it should not be taken out during entire course of titration) 5) Connect the cell to the conductometer and determine the initial conductance of the solution. 6) Add a small quality (say 0.5 ml) of standardized NH4OH solution from the burette at a time. Stir the solution well and note the new conductance value. 7) Repeat the step No.6 every time adding 0.5 ml of NH4OH solution until the conductance value shows sudden increases. Take 5 to 6 readings more after this sudden increase. 8) Plot the graphs of conductance against volume of NH4OH solution added. Determine the end point of titration Observations: 1) End point of HCl Vs NH4OH titration= X ml. 2) Volume of the diluted acid titrated = 10 ml 3) Equivalent weight of Boric acid H3BO3 = 61.84 g 4) Volume of NH4OH solution required (VA)=_________________ml(from graph) Observation Table: Observation No Volume of NH4OH solution added in ml 1 0.0 2 0.5 3 1.0 Conductance in mhos(s) Graph: Conductance VA ml of NH4OH Calculations: Standardization of NH4OH Solution: NH4OH = N1V1 = HCl N2V2 N1x 10 = 0.1 x X ∴N1 = 0.1 x X 10 Exact Normality of NH4OH = N1=__________N Normality of H3BO3 H3BO3 = N3V3 Normality of H3BO3 NH4OH = N3 = N1V1 N1X VA = 10 ___________N Amount of Boric Acid Present in given solution 1000 ml 1N NH4OH = 61.84 gm H3BO3 VA ml of N1 N NH4OH = 61.84 xVA xN1 = a= ______ gm H3BO3 1000 i.e 10 ml diluted solution contains = a gm of H3BO3 ∴ 100 ml diluted solution contains = a x 10 =b gm of H3BO3 Result:1. 2. 3. 4. Exact normality of NH4OH= N1 = _____________N Volume of NH4OH solution required (VA)=_________________ml Exact normality of H3BO3= N3 = _____________N Amount of Boric Acid Present in given solution = b= _________gm