Fatima Adam
Fatima Adam
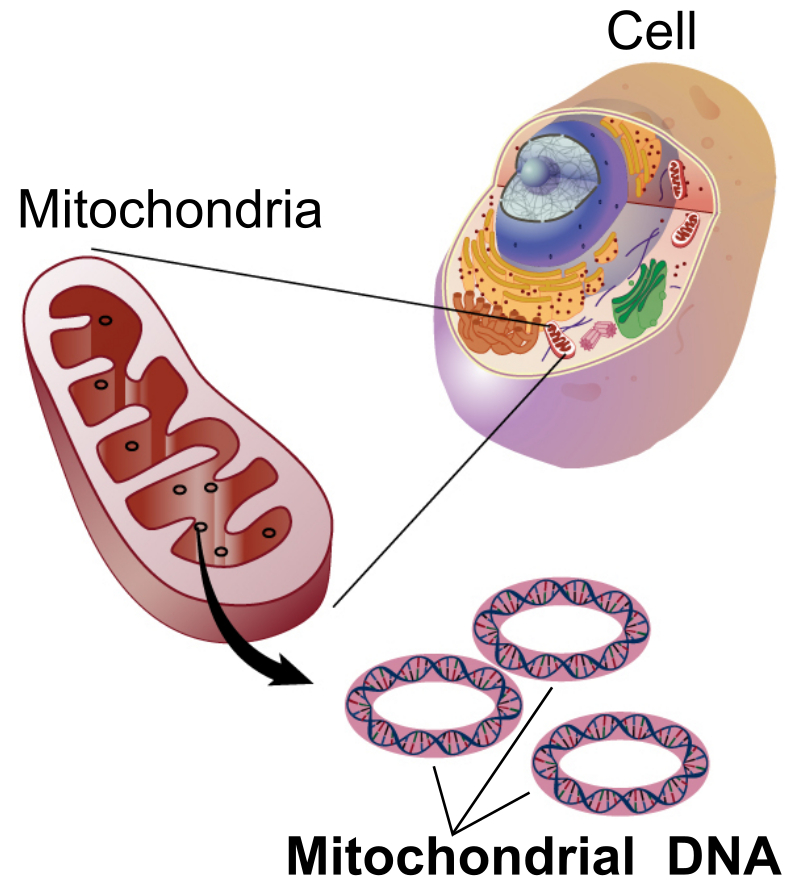
The Mitochondrial Genome (Genomics)
SDL: The Mitochondrial Genome Mitochondria are cellular organelles that have numerous essential functions including the production pf ATP by the process of oxidative phosphorylation (OXPHOS). Although most cellular DNA is found in the nucleus, the mitochondria has a small genome which encodes proteins essential for OXPHOS function. Mutations in mitochondrial proteins are the cause of mitochondrial disease, a large group of inherited metabolic disorders. This lecture will give an overview of the mitochondrial genome, how it is replicated and expressed. Diseases caused by mutations in mitochondrial proteins will be described. Learning Outcomes On successful completion of the lecture, students should be able to: Describe features of the mitochondrial genome and how it differs from the nuclear genome Explain the process of mitochondrial DNA replication Describe features and causes of mitochondrial diseases Describe diagnostic signs and tests for mitochondrial diseases Describe the genetics of mitochondrial disease, including inheritance patterns and the phenomenon of heteroplasmy Describe the process of mitochondrial replacement therapy
-
What are the features of the mitochondrial genome and how does it differ from nuclear DNA? (4)
The mitochondrial genome is circular and inherited maternally.
It contains 37 genes: 13 for proteins, 22 for tRNAs, and 2 for rRNAs.
Mitochondrial DNA is much smaller than nuclear DNA and lacks introns.
Unlike nuclear DNA, mitochondrial DNA is located in the mitochondria, not the nucleus.
-
What is the process of mitochondrial DNA replication? (3)
Mitochondrial DNA replication is semi-conservative.
It occurs in the mitochondria and is regulated by nuclear genes.
The process involves the replication of both the heavy and light strands of mitochondrial DNA.
-
What are mitochondrial diseases and their genetic causes? (3)
Mitochondrial diseases result from mutations in mitochondrial DNA or nuclear DNA affecting mitochondrial function.
Common genetic causes include mutations in the mitochondrial genome or nuclear genes encoding mitochondrial proteins.
These diseases often affect energy production and can lead to organ dysfunction, especially in high-energy tissues like muscles and the brain.
-
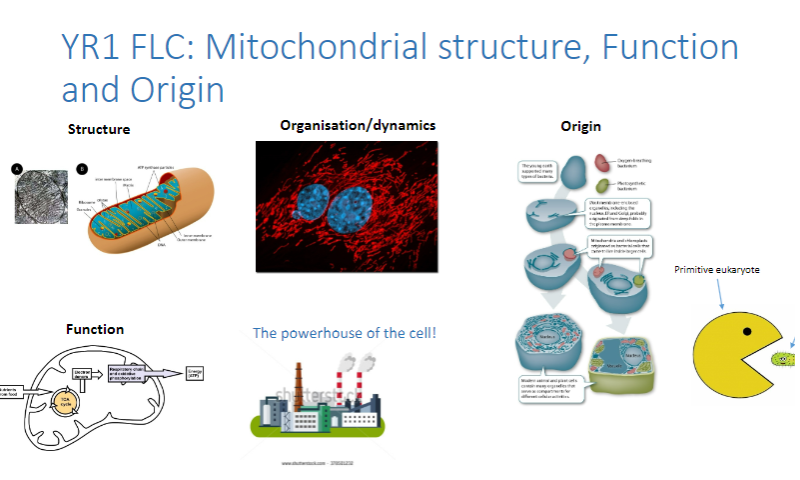
Picture demonstrating YR1 FLC: Mitochondrial structure, Function and Origin:
-
What are the features of the mitochondrial genome (mtDNA)? (7)

The mitochondrial genome is a double-stranded circular molecule (16.6kb), about 15,000 times smaller than chromosome 1.
It consists of the heavy and light strands.
It is a multicopy genome, with 10,000 to 100,000 copies per cell.
It contains 37 genes: 13 oxidative phosphorylation (OXPHOS) protein subunits, 22 transfer RNAs, and 2 ribosomal RNAs.
It lacks introns.
The D-loop is a non-coding region where replication and transcription are initiated.
Mitochondrial DNA is maternally inherited and does not undergo recombination.
-
What is the function of the mitochondrial genome in relation to oxidative phosphorylation (OXPHOS)? (2)

The mitochondrial genome encodes proteins involved in oxidative phosphorylation (OXPHOS).
OXPHOS consists of five enzyme complexes (CI-CV) that are responsible for cellular energy production.
-
What are the features of the non-coding region (NCR) in mitochondrial DNA? (3)
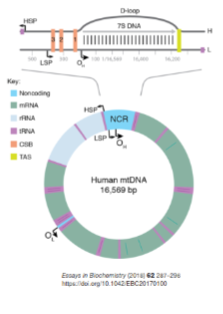
The non-coding region (NCR) contains regulatory sequences for replication and transcription.
mtDNA replication begins at the Origin of Heavy Strand (OH).
Transcription starts at the Heavy Strand Promoter (HSP) and Light Strand Promoter (LSP).
-
How is mitochondrial DNA (mtDNA) packaged into nucleoids? (3)
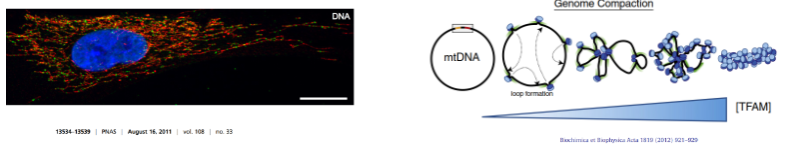
mtDNA is packaged into structures called nucleoids.
Each nucleoid contains one or two copies of mtDNA.
Transcription Factor A (TFAM) acts as a histone protein, helping to package and stabilize mtDNA.
-
What are the exceptions to the "universal" genetic code in vertebrate mitochondria? (3)
AUA and AUG both code for methionine (AUA normally codes for isoleucine in nuclear DNA).
UGA codes for tryptophan (UGA is a stop codon in nuclear DNA).
AGA and AGG are stop codons (they normally code for arginine in nuclear DNA).
-
Picture demonstrating mtDNA haplogroups:
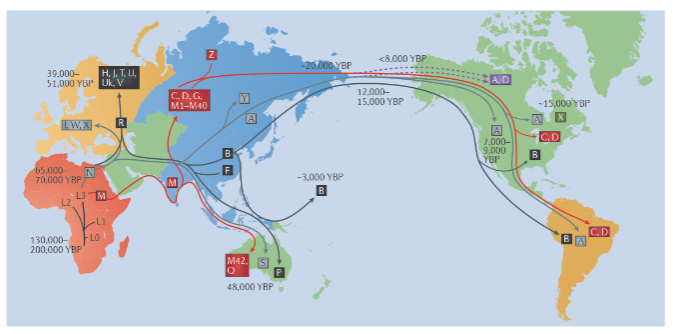
-
What is the relationship between nuclear and mitochondrial DNA in protein production? (3)
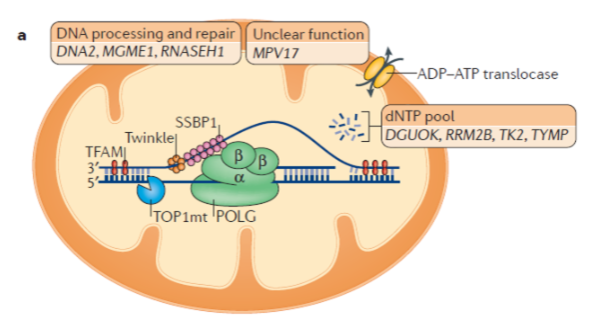
mtDNA encodes 13 proteins involved in oxidative phosphorylation (OXPHOS).
OXPHOS requires more than 100 proteins, most of which are encoded by nuclear DNA.
For mtDNA to produce these 13 proteins, it must undergo replication, transcription, and translation.
-
How are the proteins involved in replication, transcription, and translation of mtDNA produced? (2)
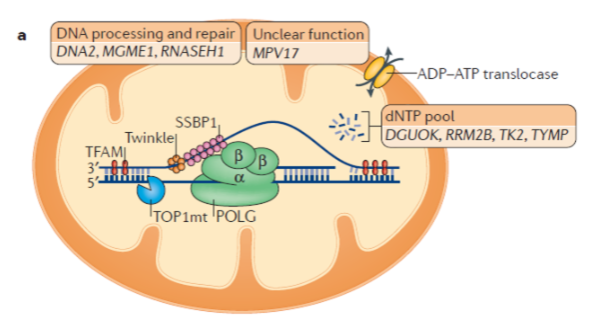
All proteins involved in replication, transcription, and translation of mtDNA are encoded by nuclear genes.
These nuclear-encoded proteins are imported into mitochondria to function.
-
How many mitochondrial proteins are there, and how are they made? (2)

In total, over 1000 mitochondrial proteins exist.
Only 13 of these proteins are made by mtDNA, while the rest are made by nuclear genes.
-
What is the role of mitochondrial DNA polymerase (Polγ)? (3)
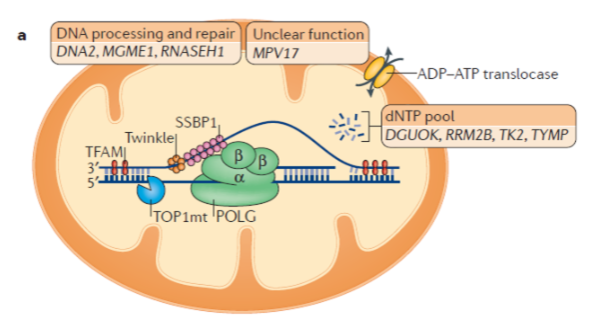
Mitochondrial DNA polymerase is a heterotrimer protein consisting of one catalytic subunit (POLγA) and two accessory subunits (POLγB).
POLγA contains a 3’ – 5’ exonuclease domain, allowing it to proofread newly synthesized DNA.
POLγB enhances interactions with the DNA template and increases the activity and processivity of POLγA.
-
What is the function of mitochondrial DNA helicase (TWINKLE)? (2)
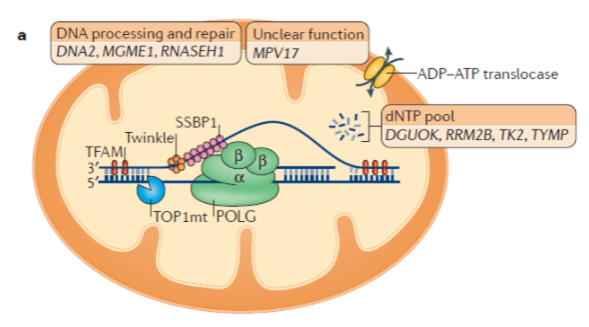
TWINKLE is a hexamer made up of six subunits.
It unwinds the double-stranded mtDNA template to allow replication by Polγ.
-
What is the function of mitochondrial single-stranded binding protein (mtSSBP)? (3)
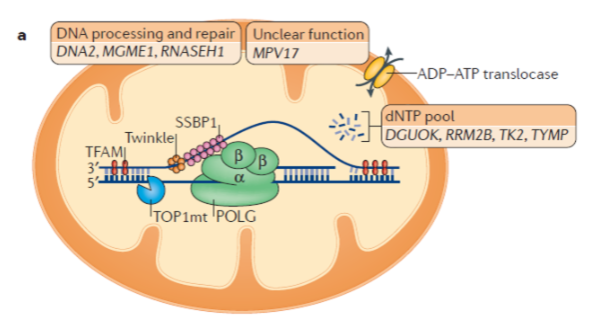
mtSSBP binds to single-stranded DNA, protecting it from nucleases.
It prevents secondary structure formation in the DNA.
mtSSBP enhances mtDNA synthesis by stimulating TWINKLE helicase activity.
-
What is the strand displacement model of mitochondrial DNA replication? (3)
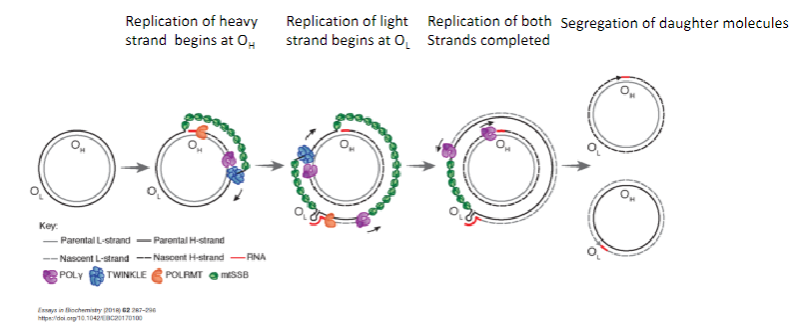
Replication of the heavy strand begins at the Origin of Heavy strand (OH).
Replication of the light strand begins at the Origin of Light strand (OL).
Segregation of daughter molecules occurs after both strands are replicated.
-
How does the strand displacement model of mtDNA replication proceed? (4)
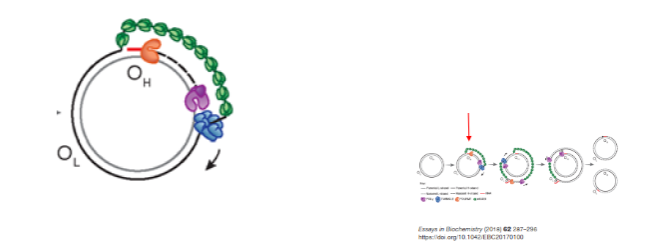
The parental heavy strand is displaced and coated with mitochondrial single-stranded binding protein (mtSSBP).
TWINKLE helicase unwinds the mtDNA.
Mitochondrial RNA polymerase (POLRMT) synthesizes an RNA primer using the light strand as a template.
POLγ uses the RNA primer to replicate DNA at the OH.
-
How does the strand displacement model of mtDNA replication proceed beyond the initial stages? (3)
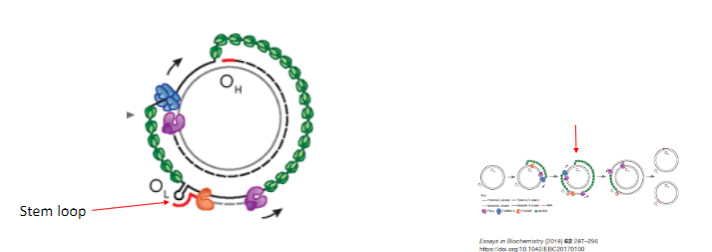
Heavy strand replication passes the Origin of Light strand (OL).
A stem-loop structure is formed, preventing mtSSBP binding.
Mitochondrial RNA Polymerase (POLRMT) synthesizes an RNA primer using the heavy strand as a template.
-
How does POLγ contribute to the strand displacement model of mtDNA replication? (2)
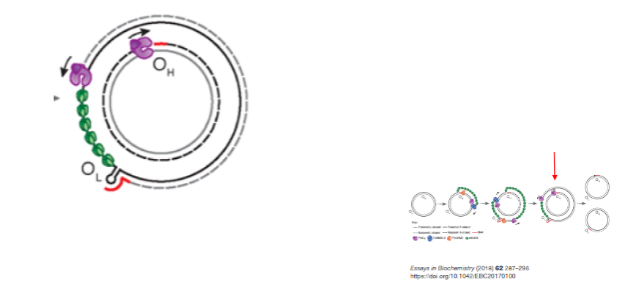
POLγ uses the RNA primer synthesized by POLRMT to replicate the light strand DNA at OL.
Synthesis proceeds until both strands are fully replicated.
-
What happens after mitochondrial DNA replication? (1)
After replication, daughter molecules are segregated.
-
What are mitochondrial diseases and their characteristics? (6)
Mitochondrial diseases are rare monogenic diseases.
They affect between 1 in 2000 and 1 in 4000 individuals.
Oxidative phosphorylation disorders are the most common form of mitochondrial disease.
These diseases often affect highly metabolic organs abundant in mitochondria.
Mitochondrial diseases can affect one (isolated) or several organ systems (multisystem).
They can start at any age and have a wide severity spectrum, ranging from adult-onset hearing loss to fatal cardiomyopathy in infancy.
-
How do mitochondrial diseases typically affect organs? (1)
Mitochondrial diseases often affect vital organs that are high in mitochondria.
-
Picture demonstrating Mitochondrial syndromes:

-
What are some clinical features and diagnostic investigations for mitochondrial disease? (6)
Clinical signs, blood and tissue histochemical/analyte measurements, neuroimaging, and DNA analysis are used for diagnosis.
Low-invasive biochemical investigations include:
Blood/CSF lactic acid >2.1 mM
Lactic acid/pyruvate ratio
Amino acids (e.g., alanine)
Organic acids
-
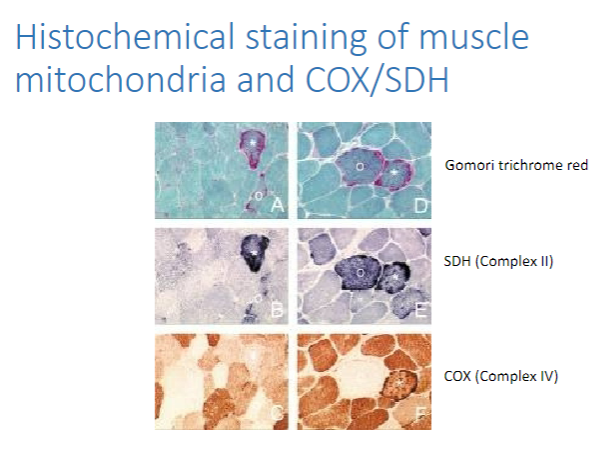
What are some muscle histology techniques used for mitochondrial disease diagnosis? (5)
Haematoxylin and eosin (H&E) staining
Gomori trichrome (to detect ragged red fibers)
SDH (to detect SDH-rich or ragged blue fibers)
COX (to detect COX-negative fibers)
Combined COX/SDH staining
-
Picture demonstrating Histochemical staining of muscle mitochondria and COX/SDH:
-
What is spectrophotometry, and how is it used to measure OXPHOS activity? (7)
Spectrophotometry is a method that measures enzyme activity by detecting changes in light absorbance during enzyme-catalyzed reactions.
It can be used on tissue or cell samples to assess the activity of various OXPHOS complexes:
Complex I
Complex II
Complex III
Complex I + IV
Complex I + III
Complex II + III
High-resolution respirometry measures oxygen consumption to provide insights into mitochondrial function.ion.
-
Picture demonstrating Blue-native gel electrophoresis of OXPHOScomplexes:
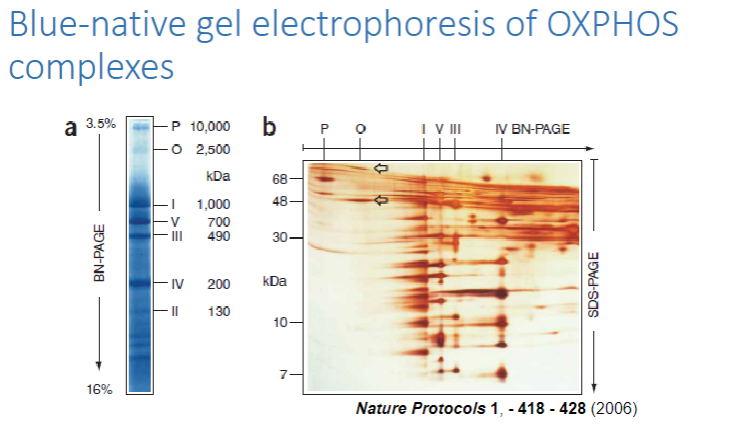
-
Picture demonstrating Electron micrography of abnormal muscle mitochondria:
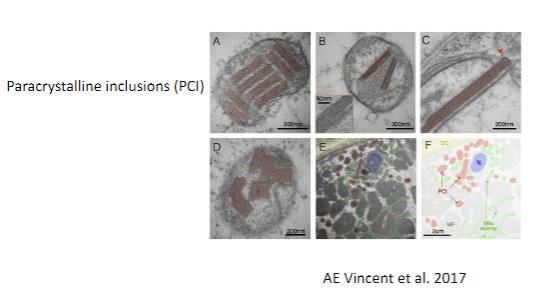
-
What are the clinical clues for diagnosing mitochondrial diseases? (7)
Characteristic syndrome or seemingly unrelated symptoms
Family history
Multiple organ involvement
Progressive nature of symptoms
Raised lactate levels
COX-negative fibers on muscle biopsy (indicating mitochondrial dysfunction)
Brain MRI changes
Genetic testing (e.g., next-generation sequencing, NGS)
-
How do mtDNA mutations cause oxidative phosphorylation disorders? (2)
mtDNA mutations lead to defects in mitochondrial function, affecting energy production.
This causes oxidative phosphorylation disorders, which impact tissues with high energy demands, such as muscles and the brain.
-
How is mitochondrial DNA disease inherited? (1)
Mitochondrial DNA diseases are maternally inherited, meaning they are passed down from mother to offspring.
-
What is heteroplasmy and how does it affect mitochondrial DNA disease manifestation? (2)
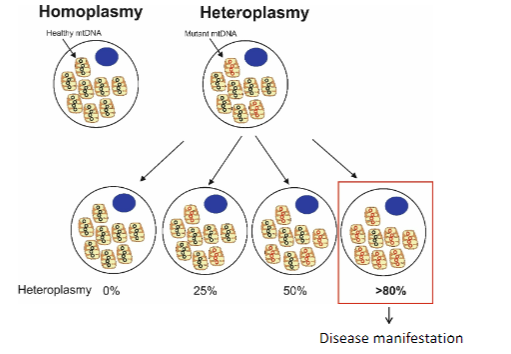
Heteroplasmy refers to the presence of a mixture of normal and mutated mitochondrial DNA within a cell.
The levels of heteroplasmy (ratio of normal to mutated mtDNA) determine the severity and manifestation of mitochondrial diseases.
-
How do heteroplasmy levels (mutation load) determine disease manifestation? (2)
Heteroplasmy levels refer to the proportion of mutated mitochondrial DNA in relation to normal mitochondrial DNA within a cell.
Higher mutation load often leads to more severe disease manifestations, as more cells are affected by dysfunctional mitochondria.
-
How is mtDNA mutation load inherited? (1)
The inheritance of mutation load is random in mitochondrial DNA, meaning that the distribution of mutated vs. normal mtDNA in offspring can vary.
-
How do heteroplasmic and homoplasmic mutations differ in mtDNA inheritance? (2)
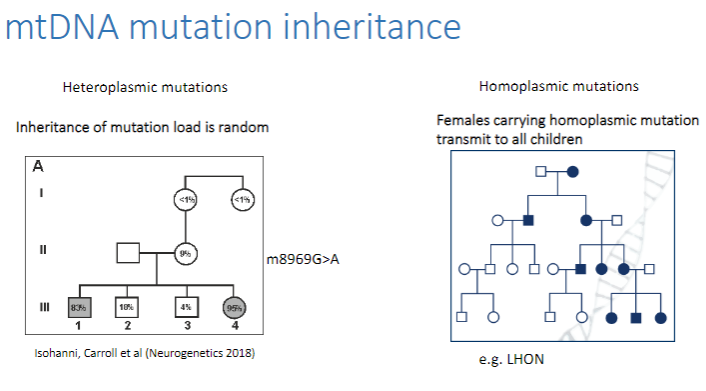
Heteroplasmic mutations: Mutated mtDNA is mixed with normal mtDNA in cells. The mutation load is random, so the transmission can vary.
Homoplasmic mutations: All mtDNA copies in a cell are mutated.
Females carrying a homoplasmic mutation transmit the mutation to all their children, as the mutation is present in all copies of their mtDNA.
-
How do heteroplasmy levels affect the penetrance and severity of disease? (3)
m8993T>G mutation:
>90% heteroplasmy = Leigh syndrome
60-75% heteroplasmy = NARP (Neurogenic muscle weakness, ataxia, and retinitis pigmentosa)
<60% heteroplasmy = Asymptomatic
Heteroplasmy levels directly influence the penetrance (likelihood of showing symptoms) and severity of mitochondrial diseases.
-
How does variable penetrance affect homoplasmic mutations? (2)
In homoplasmic mutations (100% mutated mtDNA in cells), variable penetrance still exists:
LHON (Leber's Hereditary Optic Neuropathy)
50% of affected males
10% of affected females
-
What is the mitochondrial bottleneck during oogenesis? (2)
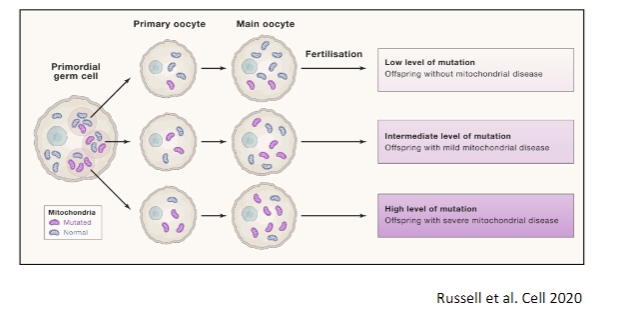
The mitochondrial bottleneck occurs during oogenesis, the formation of eggs, where a reduction in the number of mitochondria happens in early oocyte development.
This results in a limited amount of mtDNA being passed on to the next generation, which affects the heteroplasmy levels and mutation load in offspring.
-
What are the methods used for mtDNA genome sequencing? (3)
Sample types: Blood, urine, fibroblasts, or tissue.
Next-generation sequencing (NGS): Provides increased reliability and sensitivity.
Benefits: More accurate detection of low-level heteroplasmy.
-
When are muscle and liver samples necessary in mtDNA genome sequencing? (2)
Tissue-specific mutations (e.g., A3243G MELAS/MIDD mutation) may not be present in blood.
Muscle and liver tissue may be required for accurate detection of these mutations.
-
What types of mutations can be detected through mtDNA genome sequencing? (2)
Detection of single nucleotide variations (SNVs), single or multiple deletions, and duplications.
Quantitative PCR can be used to assess mtDNA depletion caused by mutations in nuclear genes that affect mtDNA replication/maintenance.
-
How can mtDNA mutations be identified in whole-exome sequencing? (2)

mtDNA mutations can be identified by "off-target" reads from whole-exome sequencing (WES).
The entire length of mtDNA is covered with several hundred short-reads, making this method highly reliable.
-
What are the advantages of using NGS for mtDNA mutation detection? (3)

No region of mtDNA is missing, unlike the nuclear genome.
The method is highly reliable due to extensive coverage of mtDNA.
Mutation load (heteroplasmy) can be accurately quantified using next-generation sequencing (NGS).
-
What nuclear genetic causes can lead to OXPHOS diseases? (2)
Mutations in mtDNA replication machinery can cause secondary mutations in mtDNA.
These mutations can result in mtDNA deletions and mtDNA depletion.
-
Where do mtDNA replication machinery mutations typically occur in the body? (4)
These mutations typically affect post-mitotic tissues, such as:
Brain
Muscle
Heart
Liver
-
What do dominant mutations in TWINKLE cause? (3)
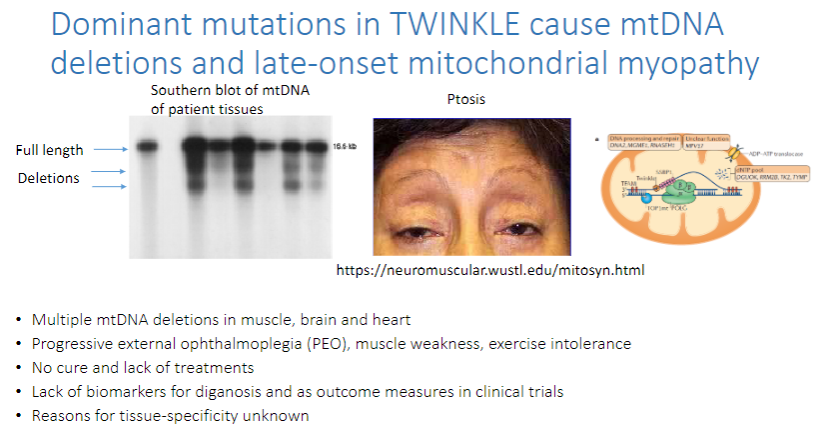
mtDNA deletions and late-onset mitochondrial myopathy.
These mutations lead to multiple mtDNA deletions in the muscle, brain, and heart.
Progressive external ophthalmoplegia (PEO), muscle weakness, and exercise intolerance are common symptoms.
-
What are the challenges in treating TWINKLE-related mtDNA mutations? (3)
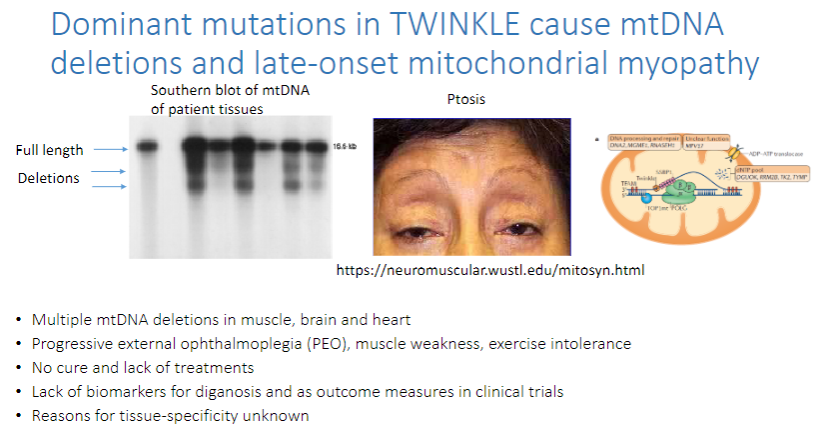
There is no cure and lack of treatments.
There is a lack of biomarkers for diagnosis and as outcome measures in clinical trials.
The reasons for tissue-specificity of the disease are unknown.
-
What is the history of mtDNA-related discoveries and sequencing? (6)
1959-62: Luft's disease recognized.
1961: Chemiosmotic theory proposed.
1963: Discovery of mtDNA.
1981: Human mtDNA sequenced.
1988: mtDNA disease mutations identified.
1995: Nuclear gene mutations discovered.
2009: Whole exome sequencing introduced.
2010: 75 nuclear genes linked to mitochondrial disease.
2020: More advanced research continued.
-
What are the approaches used for nuclear gene sequencing? (4)
Sequencing panels of disease-specific genes: Fewer genes with better sequence coverage, good for clinical practice.
Whole exome sequencing: Covers all coding genes, but some important regions may not be well sequenced.
Optimized whole exome sequencing: Captures all known disease-associated regions but costs more.
Whole genome sequencing: Sequencing everything but costly with complex data analysis.
-
What are the challenges in genetic counseling for mtDNA mutations? (4)
Homoplasmic or heteroplasmic? Affects prognosis and mutation severity.
Prognosis is difficult to predict due to mutation type, heteroplasmy levels, and variable penetrance of homoplasmic mutations.
Recurrence risks are influenced by maternal inheritance (paternal inheritance is now possible).
Homoplasmic mutations are passed to all children, while heteroplasmic mutations are passed in variable amounts.
-
What methods can be used to prevent the transmission of mtDNA mutations? (4)
Oocyte donation: Replaces faulty mitochondria.
Prenatal diagnosis: Detects mutations early in pregnancy.
Preimplantation genetic diagnosis (PGD): Ensures embryos are free of mtDNA mutations.
Mitochondrial replacement therapy: Also known as “three-parent babies.”
-
What are the key points in diagnosing and understanding mitochondrial diseases? (7)
Mitochondrial diseases are caused by mutations in mitochondrial or nuclear genes.
mtDNA mutations can be homoplasmic or heteroplasmic.
Variable penetrance occurs in homoplasmic mutations.
Heteroplasmy levels affect disease prognosis.
Nuclear gene mutations involve about 400 genes.
Genetic diagnosis methods include targeted mtDNA sequencing, gene panels, whole exome sequencing, and whole genome sequencing.