Fatima Adam
Fatima Adam
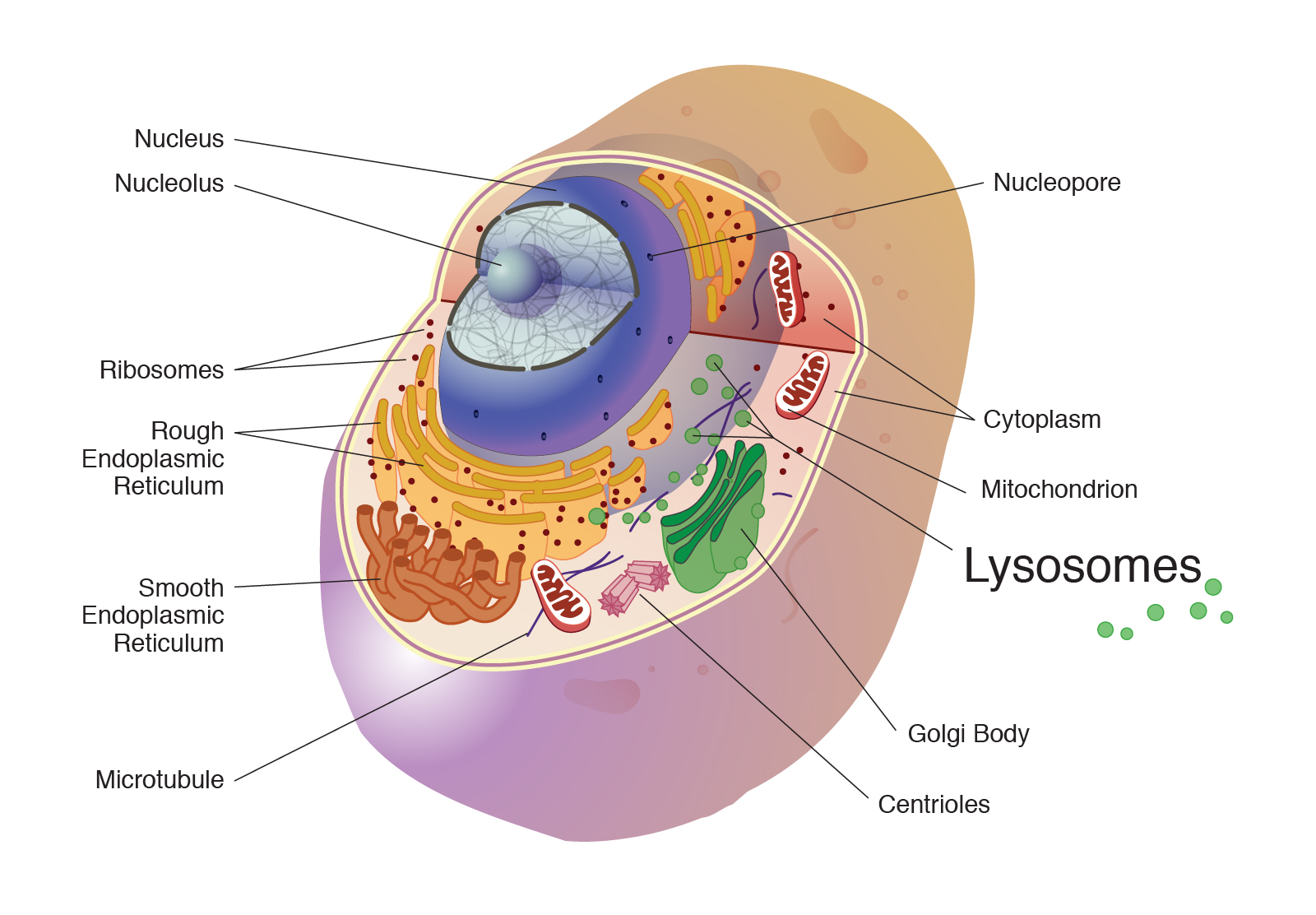
Lysosomes (The Living Cell)
The contents of lysosomes are enclosed within a single membrane and, in contrast to mitochondria, chloroplasts and the nucleus, do not contain DNA. Lysosomes (and peroxisomes) are thought to have evolved, principally, because they carry out processes that would otherwise be harmful to the rest of the cell. Lysosomes are digestive organelles containing numerous hydrolytic enzymes that can degrade material brought into them through phagocytosis, endocytosis or autophagy. Lysosomes are connected, through vesicular trafficking, to the trans-Golgi and to late endosomes. Following their synthesis in the ER, the hydrolytic enzymes are directed to lysosomes by a specific targeting signal, mannose-6-phosphate, which is added during their passage through the Golgi. Mutations in lysosomal enzymes cause lysosomal storage diseases which usually have lethal pathogenic consequences because of the build-up of undegraded material within cells.
-
How many lysosomes are there per cell?
~100
-
Do Lysosomes have a single membrane or double?
Single
-
How would you describe the contents of the lysosome?
Heterogenous (contents of lysosomes are diverse and varied in terms of the types of materials they can break down)
-
What is the pH of the lysosome?
( ̄▽ ̄*)ゞ Acidic
pH 4-5
-
How many hydrolytic enzymes does it contain?
40
-
Picture demonstrating the various functions and structures of the lysosome:
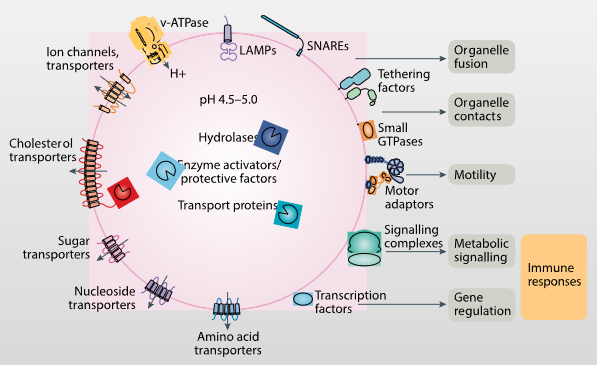
-
What does LROs stand for?
Lysosome related organelles
-
Name some cells that contain LROs and what are those LROs?
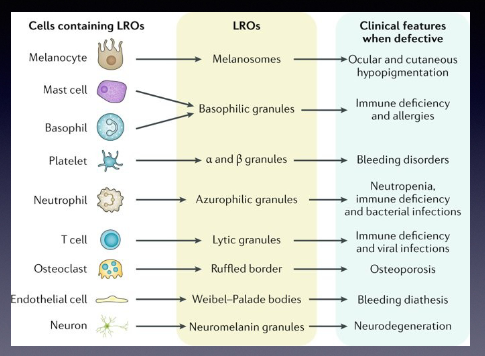
-
Picture of melanosomes at a glance:
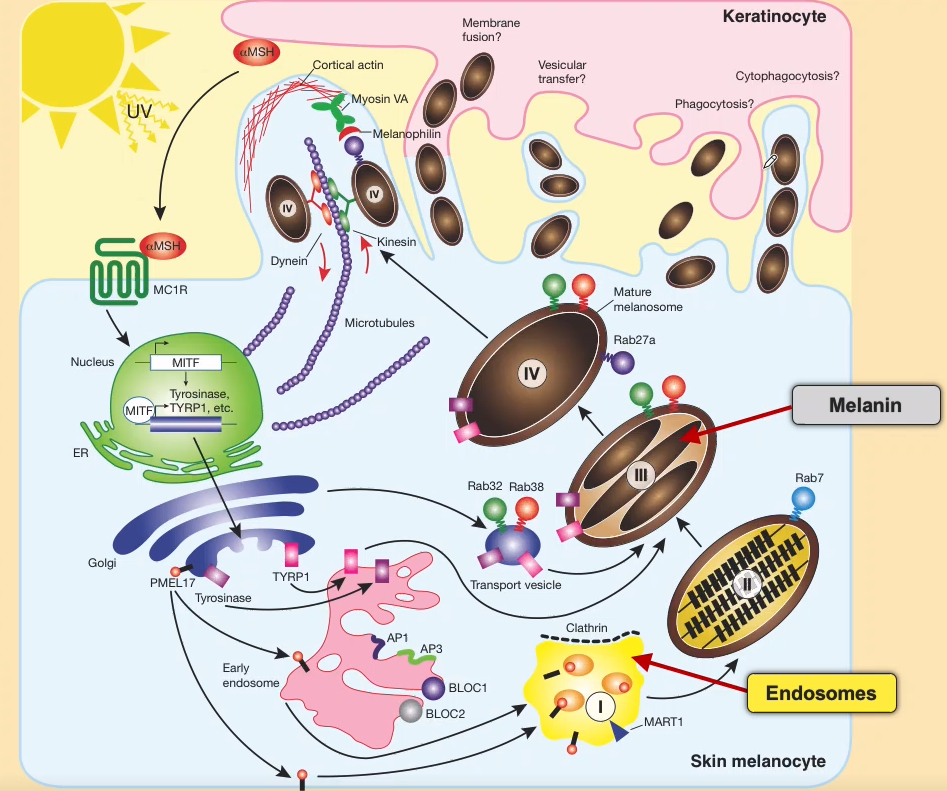
-
Where are melanosomes produced, and where are they sent off to?
-Produced in the skin melanocyte
-Sent of to the keratinocyte
-
What is the function of the lysosomes? (4)
Intracellular Digestion:
Breakdown of macromolecules (proteins, nucleic acids, lipids, and carbohydrates) using hydrolytic enzymes.
Cellular Defence:
Participate in the digestion of engulfed particles and microbial invaders during phagocytosis.
Cellular Homeostasis:
Removal and recycling of obsolete or malfunctioning cellular components, through autophagy
Programmed Cell Death (Apoptosis):
Release enzymes during apoptosis for controlled breakdown of cellular structures.
-
How are extracellular substrates delivered to the lysosomes?
• Fluid-phase endocytosis of molecules and lipoproteins (includes receptor-mediated endocytosis)
• Phagocytosis of particles ≥ 0.5 μm
-
How are intracellular substrates delivered to the lysosomes?
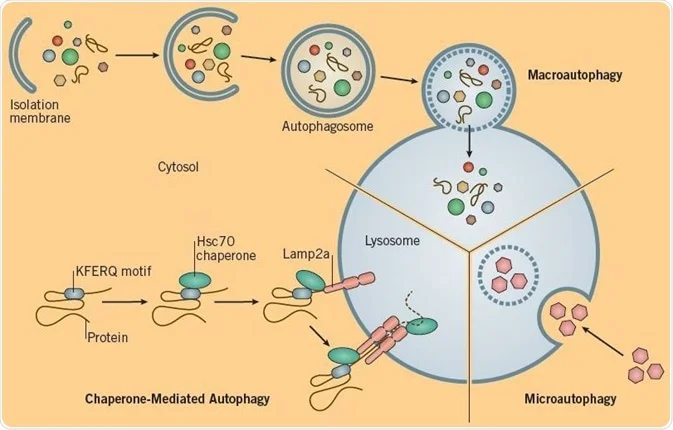
• Microautophagy (invagination/turning inward to create a pocket of the lysosomal membrane)
• Macroautophagy (cytosol or organelles wrapped in ER membrane, which then fuses with lysosomes)
• Selective transport of proteins across the lysosomal membrane
-
What does LDL stand for?
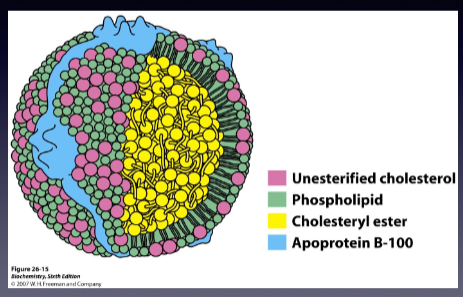
Low-density Lipoprotein
-
Outline the steps of receptor mediated endocytosis of Low-density Lipoprotein (LDL) (10)
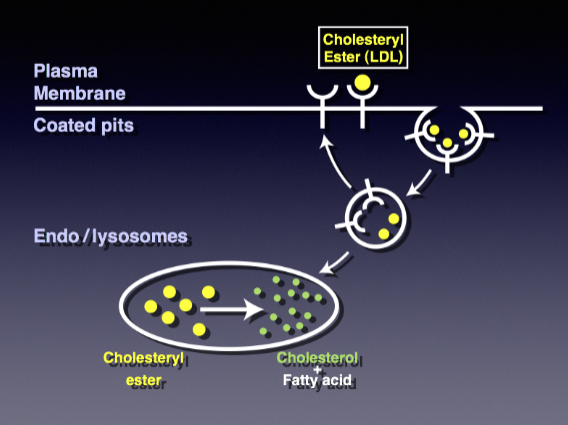
-LDL binds to cell membrane LDL receptors
-Clathrin-coated pits form on the membrane
-Pits invaginate, creating vesicles
-Dynamin aids vesicle scission from the membrane
-Shedding of clathrin coat forms uncoated endosomes
-Acidification releases LDL from receptors.
-Endosomes mature into late endosomes
-Late endosomes fuse with lysosomes.
-LDL is degraded within endolysosomes
-Some receptors recycle to the cell membrane
-
Is it true that many viruses emerge from endosomes?
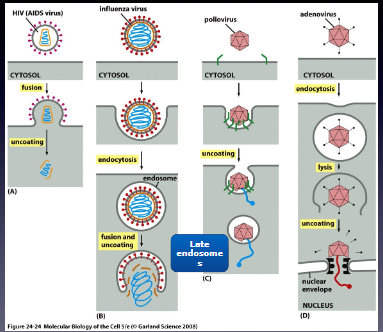
Yes!
-
Lysosomal storage disorders
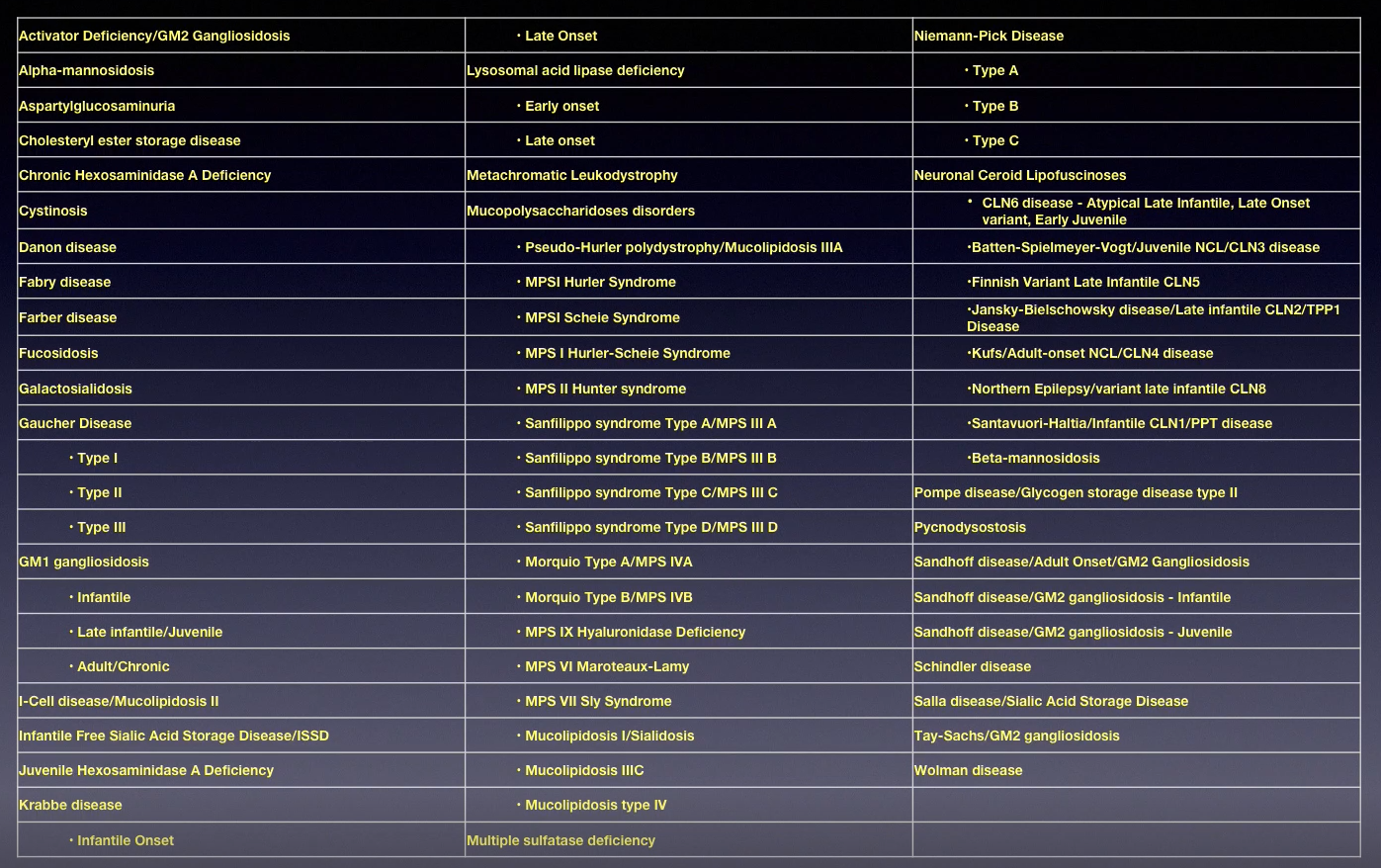
-
What do mutations in lysosomal hydrolases cause?
Substrate accumulation
-
What is the major route for targeting lysosomal enzymes to lysosomes?
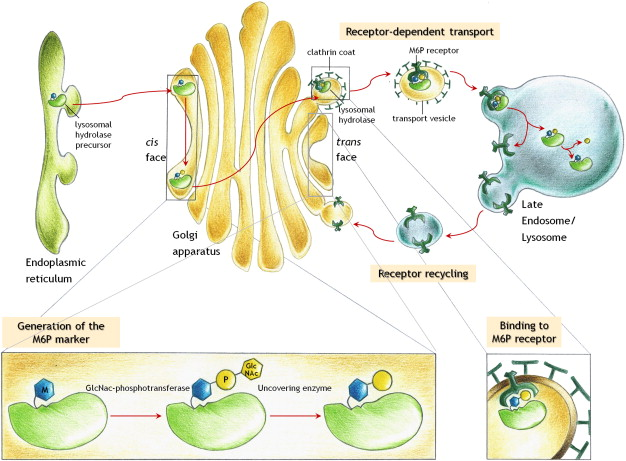
The mannose 6-phosphate (M6P) pathway
-
Outline the steps in the mannose 6-phosphate pathway (10)
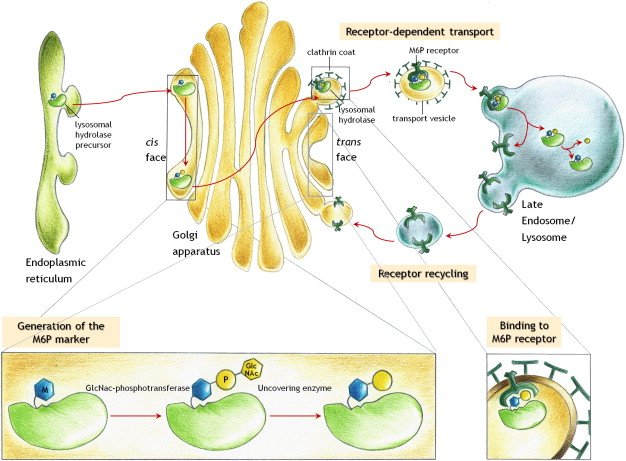
-Lysosomal enzymes made in the ER with a signal sequence
-Enzymes tagged with M6P in the Golgi
-M6P-modified enzymes bind to M6P receptors
-Clathrin-coated vesicles form with the receptor-enzyme complex
-Vesicles transport enzyme-receptor complexes to endosomes
-M6P receptors recycled to the Golgi.
-Endosomes mature into late endosomes
-Late endosomes fuse with lysosomes
-Enzymes released into lysosomes for degradation.
-M6P pathway regulates lysosomal enzyme levels
-
Mutations in the GNPTG gene cause what?
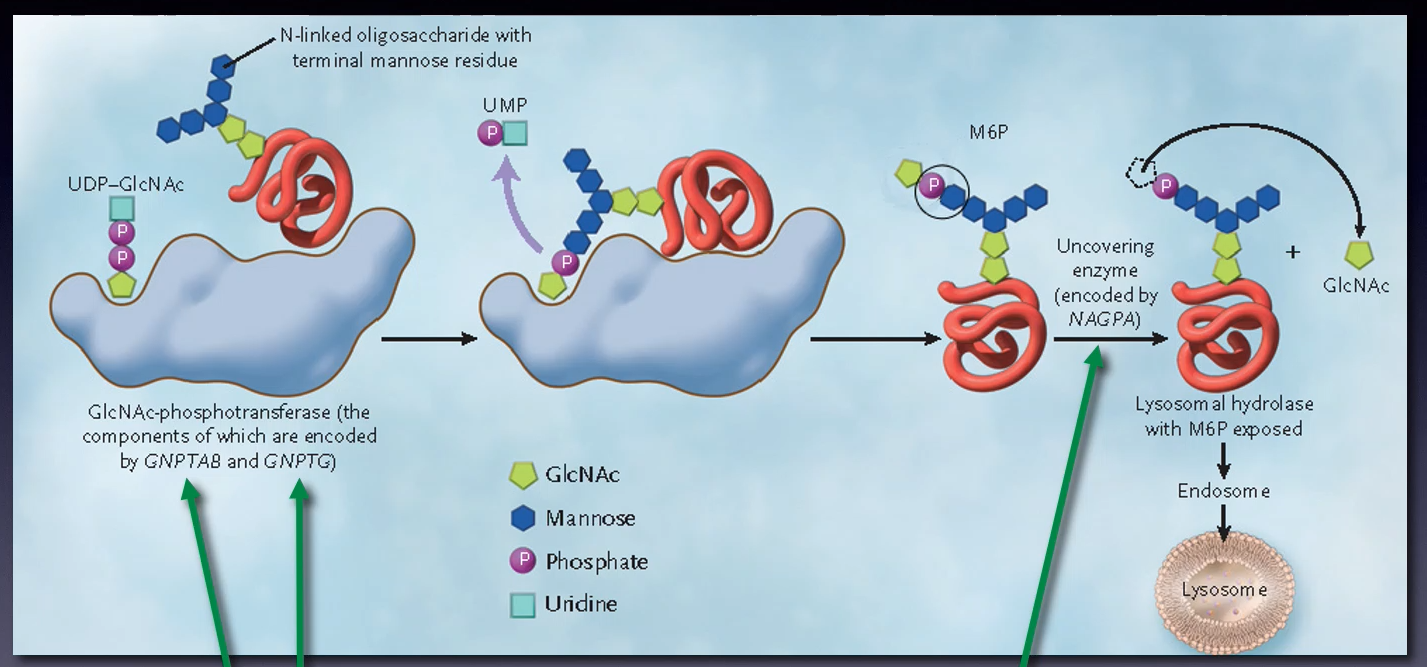
Mucolipidosis type III (Pseudo-Hurler polydystrophy)
-
Mutations in the GNPTAB gene cause what?
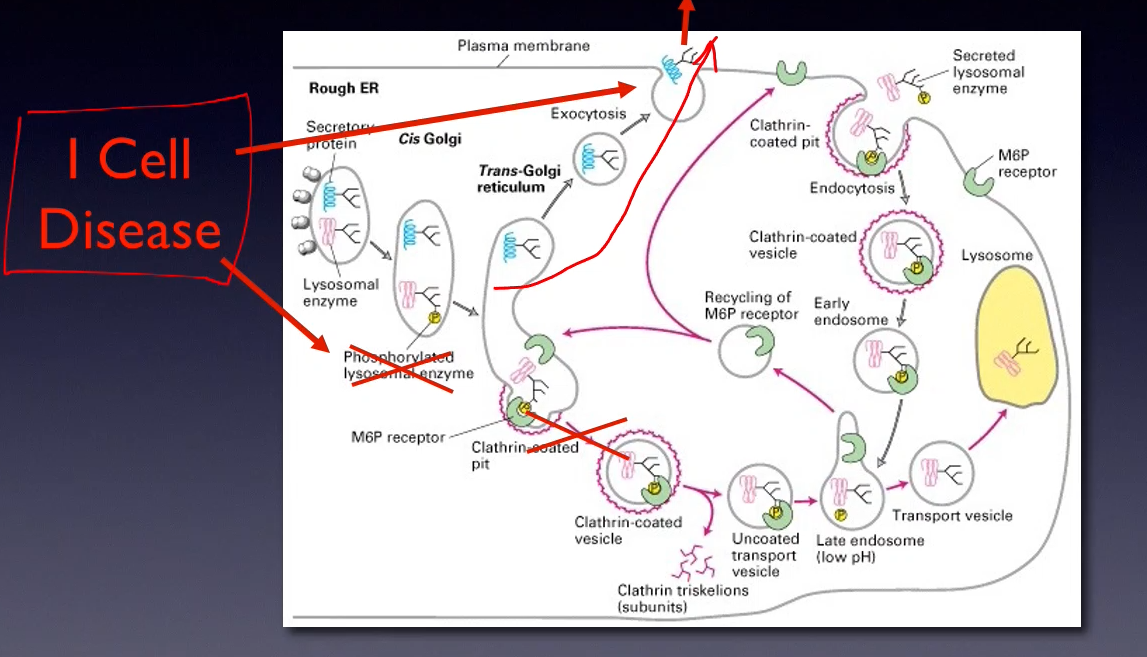
Mucolipidosis type II (I-cell disease) or type III
-
Mutations in the genes GNTAB, GNPTG or NAGPA can also cause what?
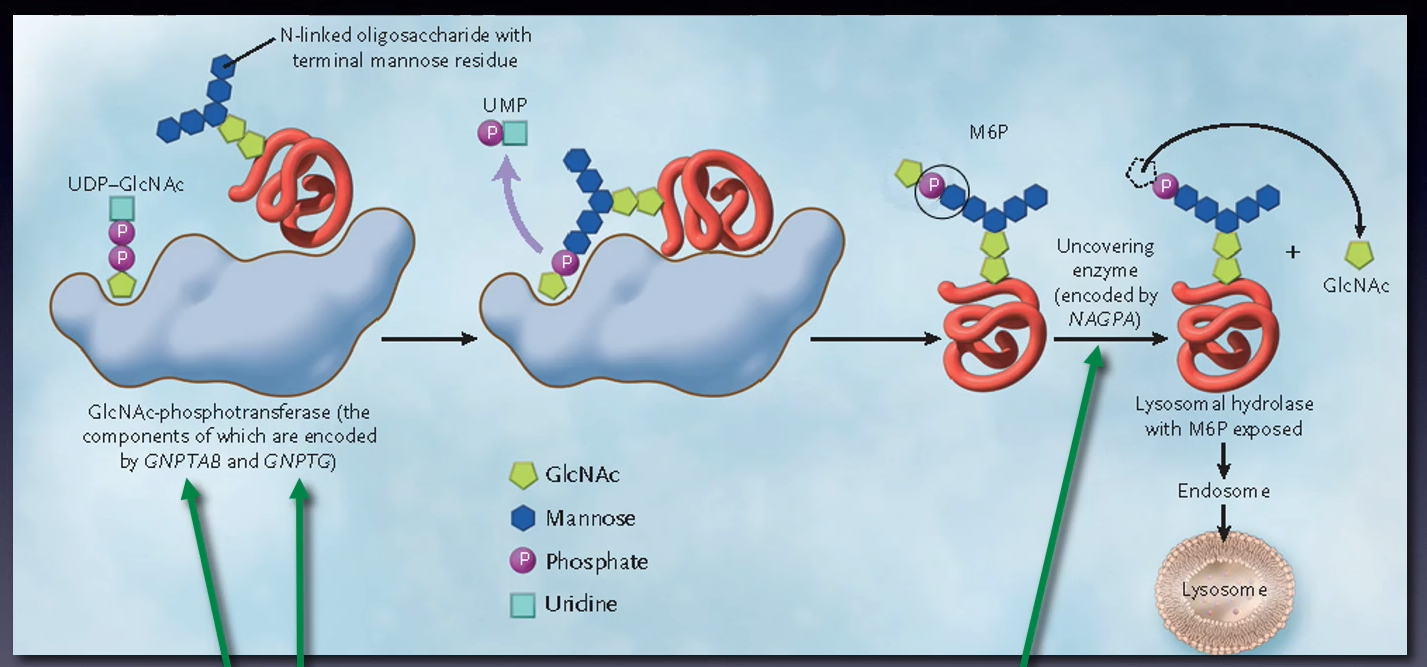
Persistent stuttering
-
I-Cell disease (Mucolipidosis type II) causes several problems, name them (6)
-Skeletal abnormalities
-Developmental delay
-Enlarged liver and spleen
-Impaired hearing
-Death from pneumonia or congestive heart failure usually occurs within the first decade of life
-Autosomal-recessive disorder caused by a deficiency of the enzyme “GlcNAc phospho-transferase”
-
What is the role of the vacuolar ATPase?
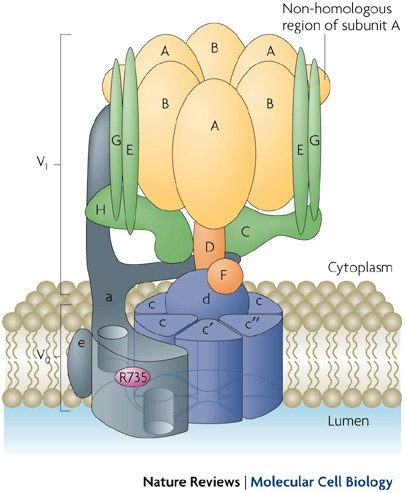
Transports protons into lysosomes and related organelles while consuming ATP