what are pharmokinetics?
what our bodies do to drugs
what is the absorption of drugs?
the drug concentration that reaches the bloodstream
what are the 3 routes of administration? what are their absorption barriers + patterns?
IV: none, instantaneous
IM/subcut: capillary wall (weak), water-soluble = rapid, poorly soluble = slow
PO: GI epithelial + capillary wall, slow + variable
what are advantages / disadvantages of IV administration?
rapid onset (emergencies!)
no 1st pass effect
allows use of large fluid volumes
expensive/inconvenient
poor self-administration
water-soluble drugs ONLY
what are advantages / disadvantages of IM/subcut administration?
allows use of poorly soluble drugs + depot preparations (large dose gets slowly absorbed)
inconvenient
possible discomfort/injury
poor self-administration
what are advantages / disadvantages of PO administration?
easy self-administration
convenient/inexpensive
variable availability (1st pass)
possible nausea/vomiting
conscious/cooperative patients ONLY
what are 3 other ways of administration that are less common?
inhalation -> rapid delivery to airways
direct injections at target -> ex: spinal cord
topical -> ointment, drops...
explain PO drug formulations
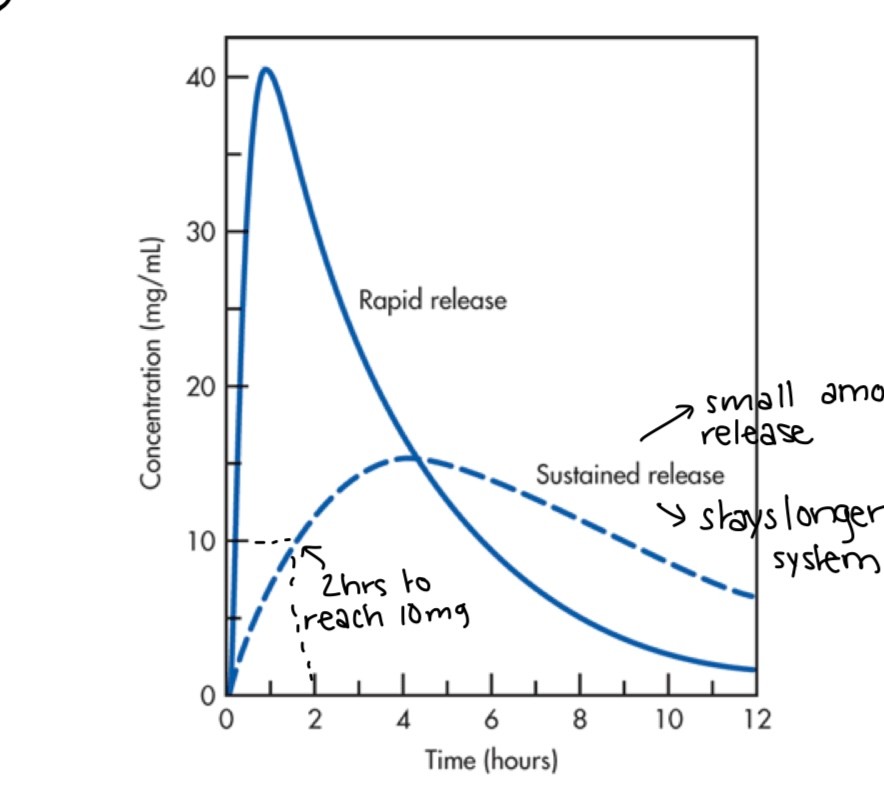
2 kinds:
1. tablets (rapid-release): as soon as taken it is released and starts to decline
2. sustained/extended release (stays longer in system): steady absorption rate, facilitates admin (less doses), but more expensive
enteric coated: protects GI mucosa + variable dissolution/absorption
what does rate/speed of absorption mean?
onset of effects
what does magnitude/amount mean?
peak effect intensity
what are the 7 factors affecting PO absorption?
lipid solubility
pH (ion trapping)
breakdown
dissolution
rate of gastric emptying (constipated = slower)
surface area
intestinal perfusion (following meal time there is increase of BF to intestine)
what is difference in drug membrane transport between water vs lipid soluble drugs?
lipid soluble -> more easily diffuse across cell membrane, go inside cells easily
water soluble -> not easy, needs help
what are 3 types of membrane transport?
simple diffusion
facilitated diffusion
primary active transport
what is P-glycoprotein/MDR-1 role?
protect our bodies from harmful chemicals that we have ingested, it pumps out cells that have "foreign"/"toxic" compounds in them
where are main locations of P-glycoprotein/MDR-1 ?
BBB, intestines, placenta, kidneys
how do P-glycoprotein/MDR-1 affect drugs?
inverse correlation with absorption
high activity = low absorption
low activity = high absorption
how do strong acids + bases affect pH?
always ionized + contribute to pH
strong bases increase pH in EVERY context
strong acids decrease pH in EVERY context
remember pH affect absorption
how do weak acids + bases affect pH?
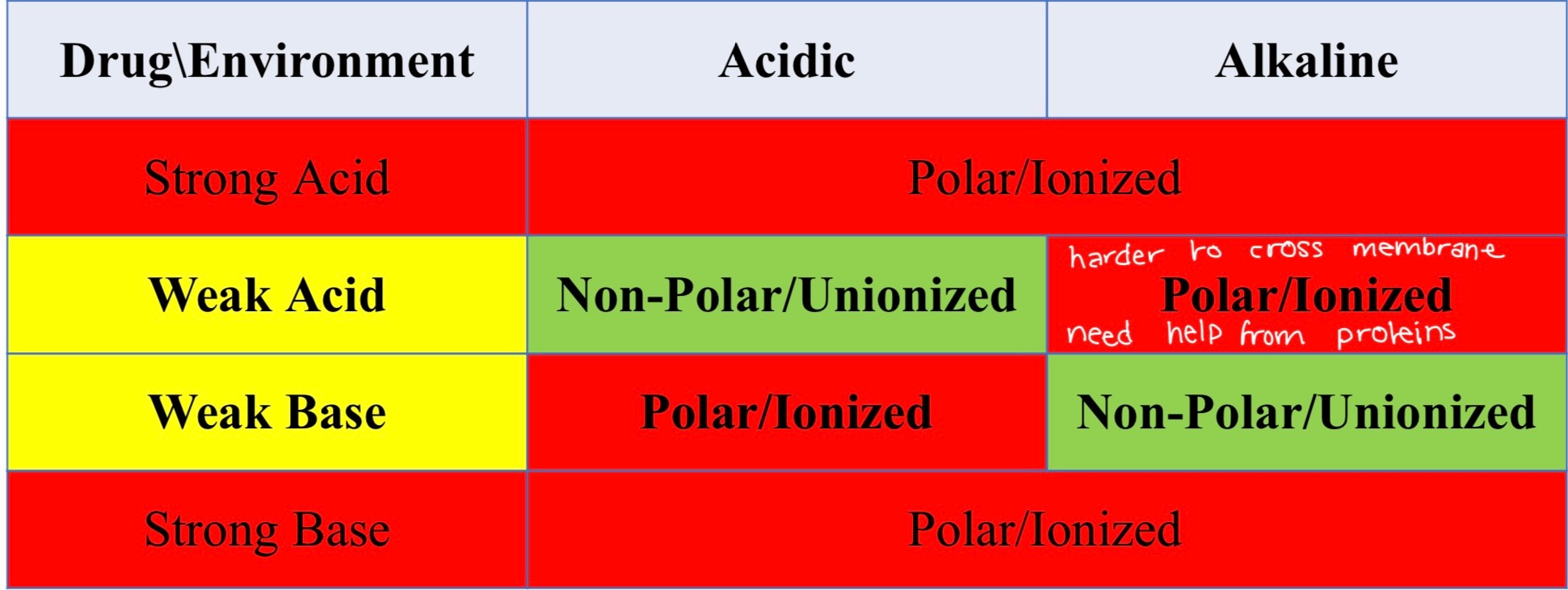
ionization depends on pH of their environment
weak bases ionized in acidic environment
weak acids ionized in alkaline environments
remember pH affect absorption
what is ion trapping? give example
drug accumulation on side of membrane when polar
ex: aspirin (weak acid) accumulates more in plasma side
what is the distribution of drugs?
movement of drug to cells + tissue
explain tissue perfusion + exit
once drug is in bloodstream, it has to come out to reach organs
lipid-soluble drugs get out easily, polar drug needs a bigger pore
if drug gets bound to plasma protein, it cannot exit bloodstream
the amount of blood pumped to different organs is not the same for every organ, certain organs get more drugs
ex: kidneys get 22% blood, while heart muscle gets 3%
how do plasma proteins affect drugs?
albumin-bound drugs get trapped in bloodstream
what are the major determinants of how plasma proteins affect drugs?
albumin levels -> synthesized by liver, so depends on liver health
drug-albumin affinity -> not all drugs bind to albumin, ex: warfarin has 97% albumin affinity (if you give 100g, 97g will bind to albumin)
how can you compare albumin bound drugs to sports?
are like sports players on the bench, ready to play but irrelevant at the moment
what drug concentration is important from a therapeutic effect perspective?
free drug concentration !!
how do optional barriers affect drugs?
remember factors affecting GI absorption, can be applicable here
non-polar/lipid-soluble drugs have easy access -> doesn't really affect them
drug can get trapped ex: placenta barrier
drugs that act inside cells must cross plasma membrane to enter
BBB is strong barrier -> polar drugs NEED transport system to access it, it is hard to treat brain problems
ex: alcohol/heroin/nicotine have easy access, morphine/penicillin do not
what is drug metabolism ?
alteration of drug chemical structure
what are the hepatic enzymes pharmacologic effects? where are they found?
bio activation of Rx
inactivation of Rx
promote Rx excretion
liver - some drugs are inactive and need to be activated by liver, it basically cleans up before sending to your blood
what are 2 phases of drug metabolism that occur in liver?
phase 1: consists of reduction, oxidation, or hydrolysis reactions. These reactions serve to convert lipophilic drugs into more polar molecules
phase 2: adding hydrophilic groups to the original molecule, a toxic intermediate or a nontoxic metabolite formed in phase I, that requires further transformation to increase its polarity.
NOT ALL DRUGS GO THROUGH BOTH PHASES
what is main enzyme used in drug metabolism? what is it at higher risk for
CYP3A4/5 -> higher risk of Rx interactions (patient taking multiple drugs who need this enzyme to be metabolized
what are the 4 types of metabolisers (not everyone reacts to drugs the same way)?
normal: most common/wild type allele
poor/non metabolisers: 2 loss-of-function (LoF) alleles
intermediate: 1 LoF allele
ultrarapid: gain-of-function allele or gene duplication (either hyperactive version or more doing same job)
what is the first pass effect + bioavailability ? what are its characteristics?
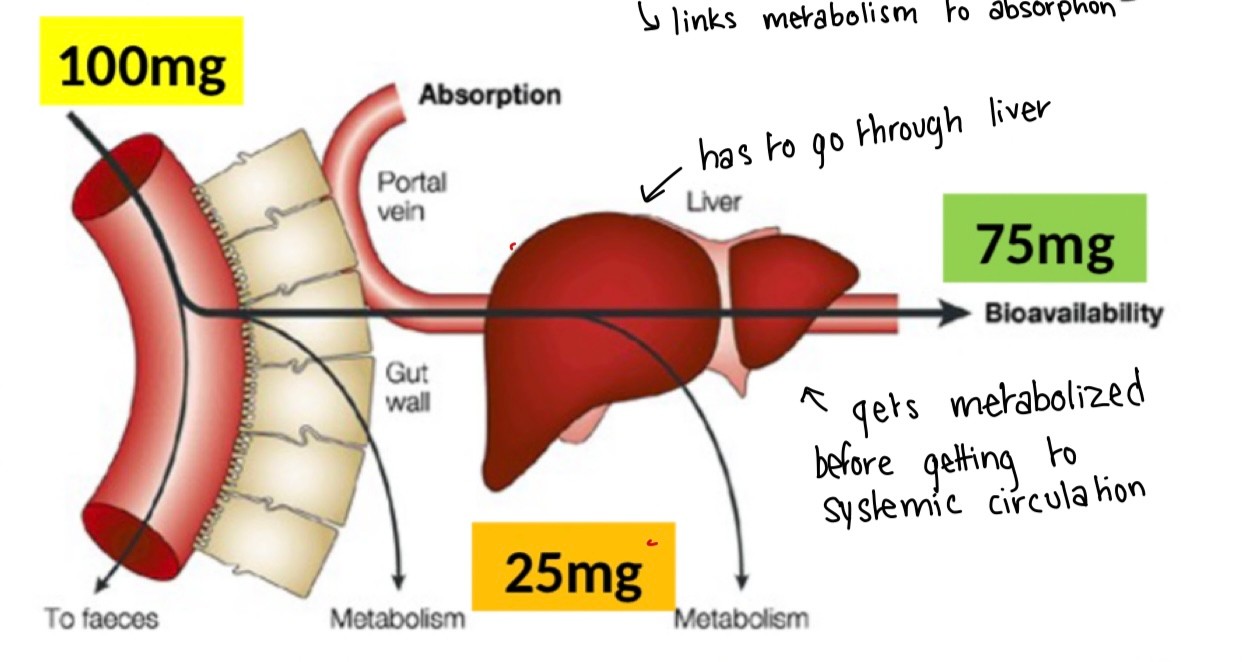
a phenomenon in which a drug gets metabolized at a specific location in the body that results in a reduced concentration of the active drug upon reaching its site of action or the systemic circulation
significant only for PO administration
weak for topical + inhalation
bypassed completely by parenteral admin
if you know optimal therapeutic effect happens when concentration is 90mg, how much should you administer PO if bioavailability is 75%?
?mg * 75% = 90 mg -> 90 * 100/75 = 120
what are 3 special metabolic considerations for nurses?
age -> effectiveness of metabolism
indication + inhibition/competitor -> alcohol increases alcohol metabolism, ritonavir inhibits CYP3A4 metabolism
nutrition
what is the enterohepatic recirculation? who is it exclusive to?
biliary excreted drug reabsorbed in intestine instead of being removed by body
drugs undergoing glucoronidation (Glu)
explain mechanism of enterohepatic recirculation
liver transferase -> drug Glu -> inactivation -> drug Glu travels to Gi tract via bile -> gut microbiome enzymes -> hydrolysis of Glu -> free drug increases and returns to circulation
what is excretion of drug?
drug removal of body
NOT elimination
where does most excretion occur? what factors affect it?
renal excretion
age
pH -> adjusting pH of urine can affect if drug is reabsorbed or not
tubular transporter competition -> water soluble drugs rely on transporters
what is equation for urinary excretion?
GF + TS - TR
GF glomerular filtration: the process that your kidneys use to filter excess fluid and waste products out of the blood into the urine collecting tubules of the kidney, so they may be eliminated from your body
TR tubular reabsorption: the kidneys reabsorb useful substances, such as glucose, amino acids, and electrolytes, from the filtrate back into the bloodstream.
TS tubular secretion: the transfer of materials from peritubular capillaries to the renal tubular lumen; it is the opposite process of reabsorption.
what are 2 phases of elimination?
phase 1: inactivation using P450 enzymes
phase 2: make it more water soluble w/transferase
Not all drugs need to go through both:
some might only need to become water soluble : 2 only
some might already be water soluble: 1 only
what are 3 types of non renal excretion?
lungs -> alcohol (10%), anaesthetics
breast milk -> mostly lipid-soluble/non polar, educate moms!
bile/feces/saliva/sweat -> therapeutically insignificant, useful for drug detection
what is a dose response curve? (DRC)
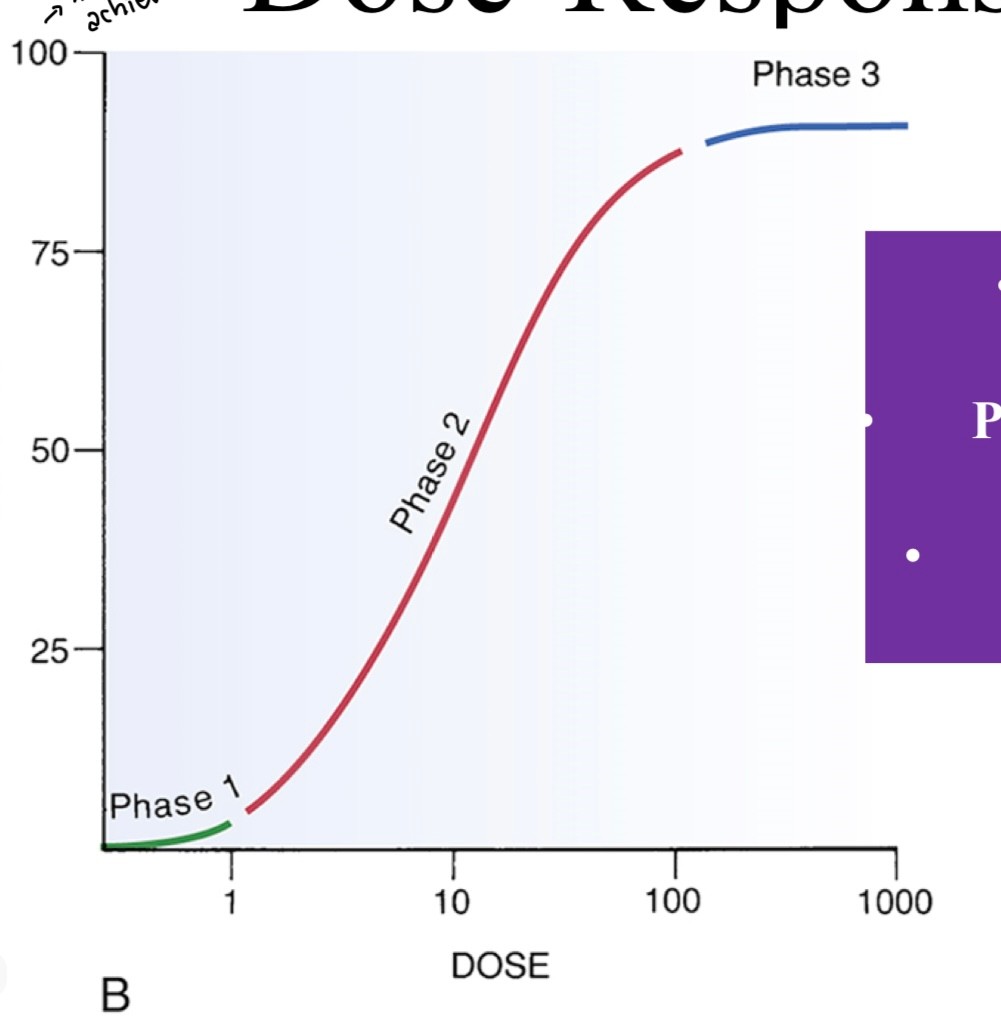
the graphical representation of the relationship between the dose of a drug versus the effects that the drug exerts on the system tested, depciting the magnitude of the response of the organism, either therapeutic or toxic
phase 2 = graded relationship (most drugs), if no phase 2 = all or nothing drugs (hypnotics)
what is efficacy vs potency of drug?
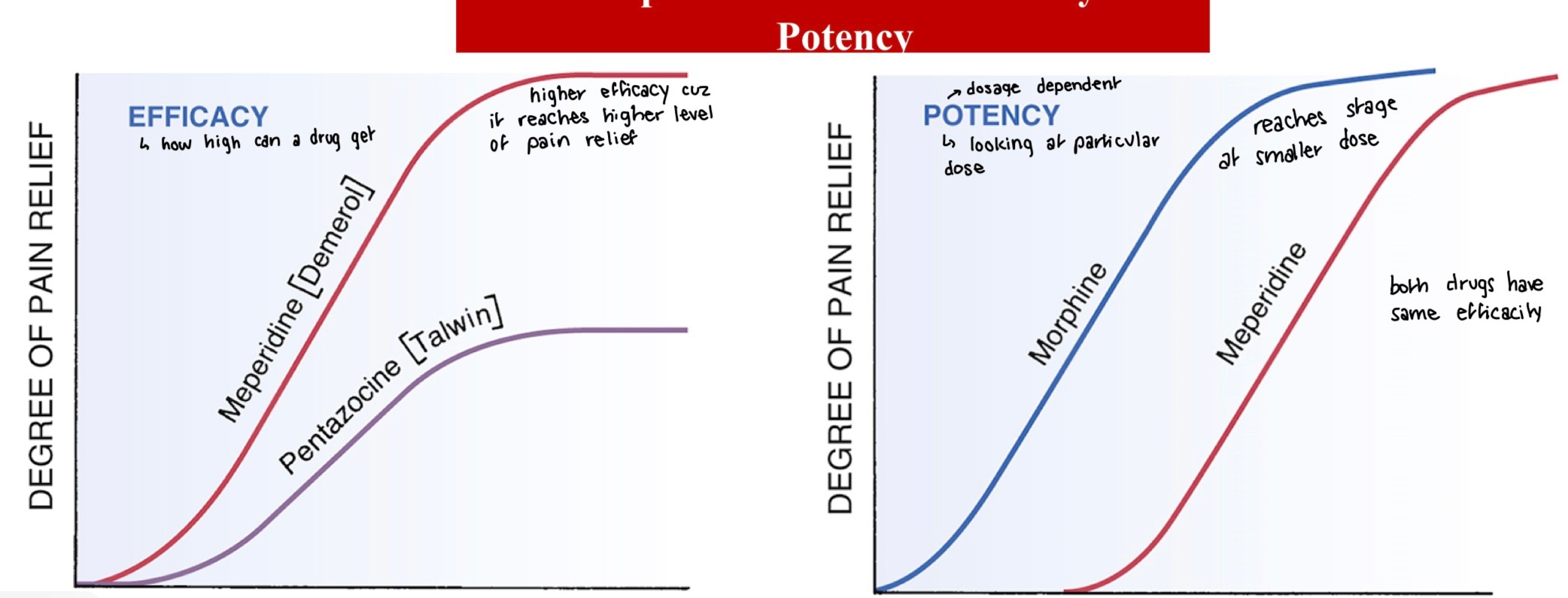
efficacy -> how high can a drug get in terms of action: if higher efficacy if higher pain relief (more important therapeutically)
potency -> dosage dependant, how much relief depending on dose, can have same efficacy but different potency
what is affinity vs efficacy of drug?
high affinity = binds easily
efficacy = how much of an effect can it have
occupation governed by affinity
activation governed by efficacy
what is drug agonist? what is drug antagonist?
a substance that mimics the actions of a neurotransmitter or hormone to produce a response when it binds to a specific receptor in the brain. affinity + efficacy
A chemical substance that binds to and blocks the activation of certain receptors on cells, preventing a biological response -> ex: naloxone is antagonist to opioids. Have affinity but 0 efficacy
what is 4 levels of agonists/antagonists ? explain with door analogy
full agonist:
if door is closed at start, opens door to max
if door is ajar, opens door to max
partial agonist:
if door is closed at start, cracks door open
if door is ajar, opens door a bit more
antagonist:
if door is closed at start, nothing changes, door remains closed
if door is ajar, nothing changes, door remains ajar
inverse antagonist:
if door closed at start, N/A
if door is ajar, closes door
what are 2 antagonists types?
noncompetitive/insurmontable: irreversible binding decreases # of available receptors -> changes shape of enzyme so it cannot bind to substrate -> rightward shift depends on relative affinity between agonist/antagonist
competitive/surmontable: reversible binding at same site -> interferes w/active site of enzyme so substrate cannot bind -> curve decline depends on # of spare receptors
what is difference between receptor selectivity vs sensitivity?
selectivity: how many different types of drugs can bind
sensitivity: how many receptors are available
what are the 4 main receptor families?
1. ligand-gated ion channels (ionotropic receptors)-> when drug binds it open/closes ion channel
2. G-protein-coupled receptors (metabotropic) -> most common for drugs
3. kinase-linked receptors -> rare, insulin
4. nuclear receptors -> hormones
not all drugs bind to receptors, what are 5 cases?
chelating agents -> physically trap chemical molecules: antacids (target H+), resins (target cholesterol), dimercaprol (target metals)
laxatives -> chemical molecule retention : magnesium sulfate (osmotic water retention in GI)
antiseptics -> precipitate bacterial proteins
protective coats -> prevent chemical cell injuries: sucralfate (coat stomach lining to prevent GI ulcers), sunscreens (coat skin cells)
anti-mitotic agents -> prevent cell cycles stages: taxanes (stabilize microtubules to inhibit cell division)
what is ED50?
does to achieve therapeutic objective in 50% of individuals, average effective dose
what is LD50? TD50?
LD50 (animal studies) lethal dose in 50% of individuals
TD50 (human studies) toxic dose in 50% of individuals
what is therapeutic index (TI)?
LD50/ED50 -> larger TI, safer drug
fun fact alcohol TI is 4
what is therapeutic window?
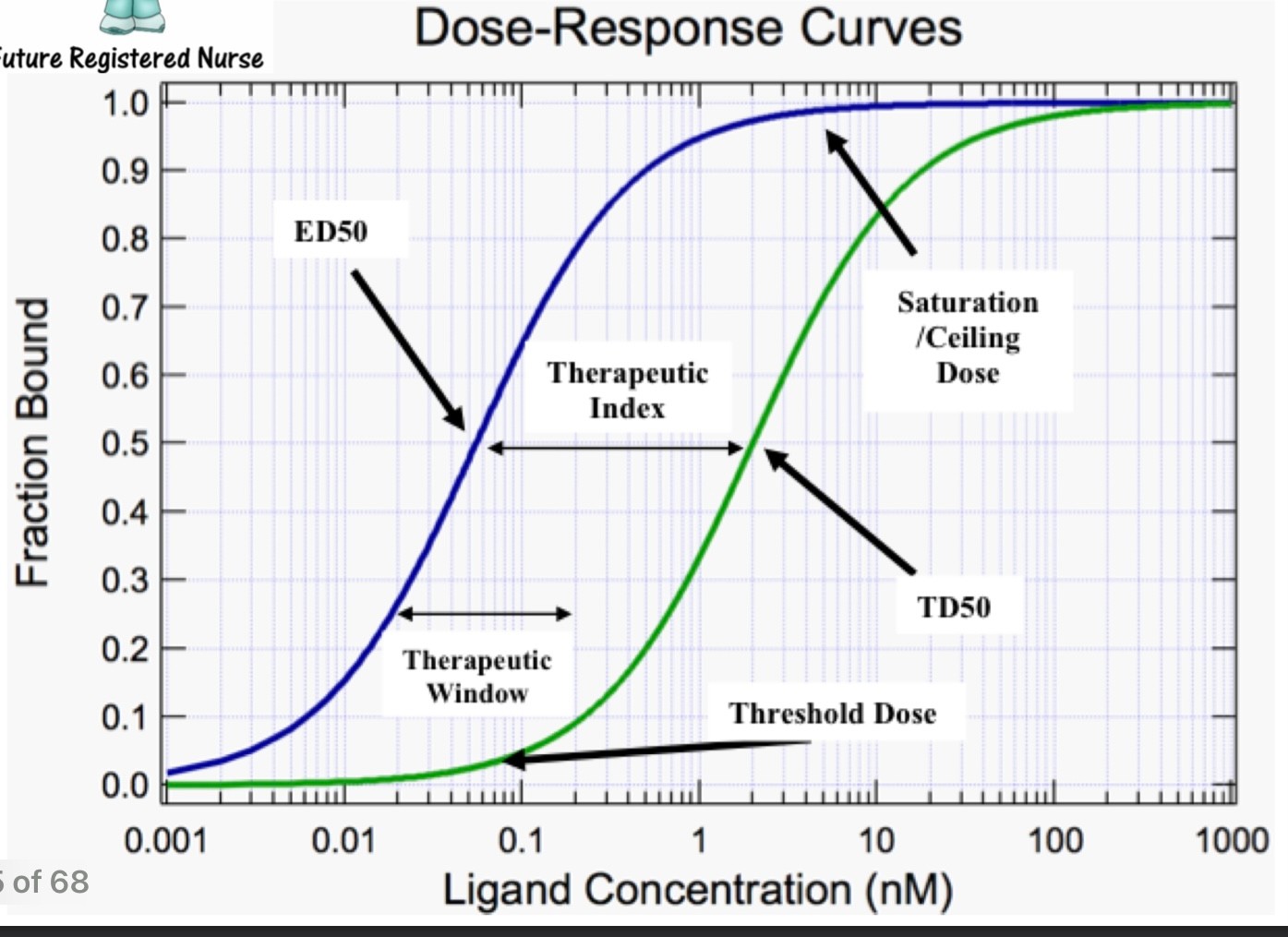
The dose range of a drug that provides safe and effective therapy with minimal adverse effects.
why do we care about plasma drug concentration?
it directly correlates w/therapeutic and toxic response
what is the "duration"?
the amount of time the drug response is within therapeutic range (beneficial effects)
what does it mean if drug has a wider therapeutic range? a narrower one?
safer drug
increased toxicity risks
what effects the "trajectory" of plasma drug concentration? how does absorption affect it? absorption, distribution, metabolism? metabolism and excretion?
kinetics
time until minimum effective concentration
peak height
duration length
what do you need to watch out for when choosing admin route?
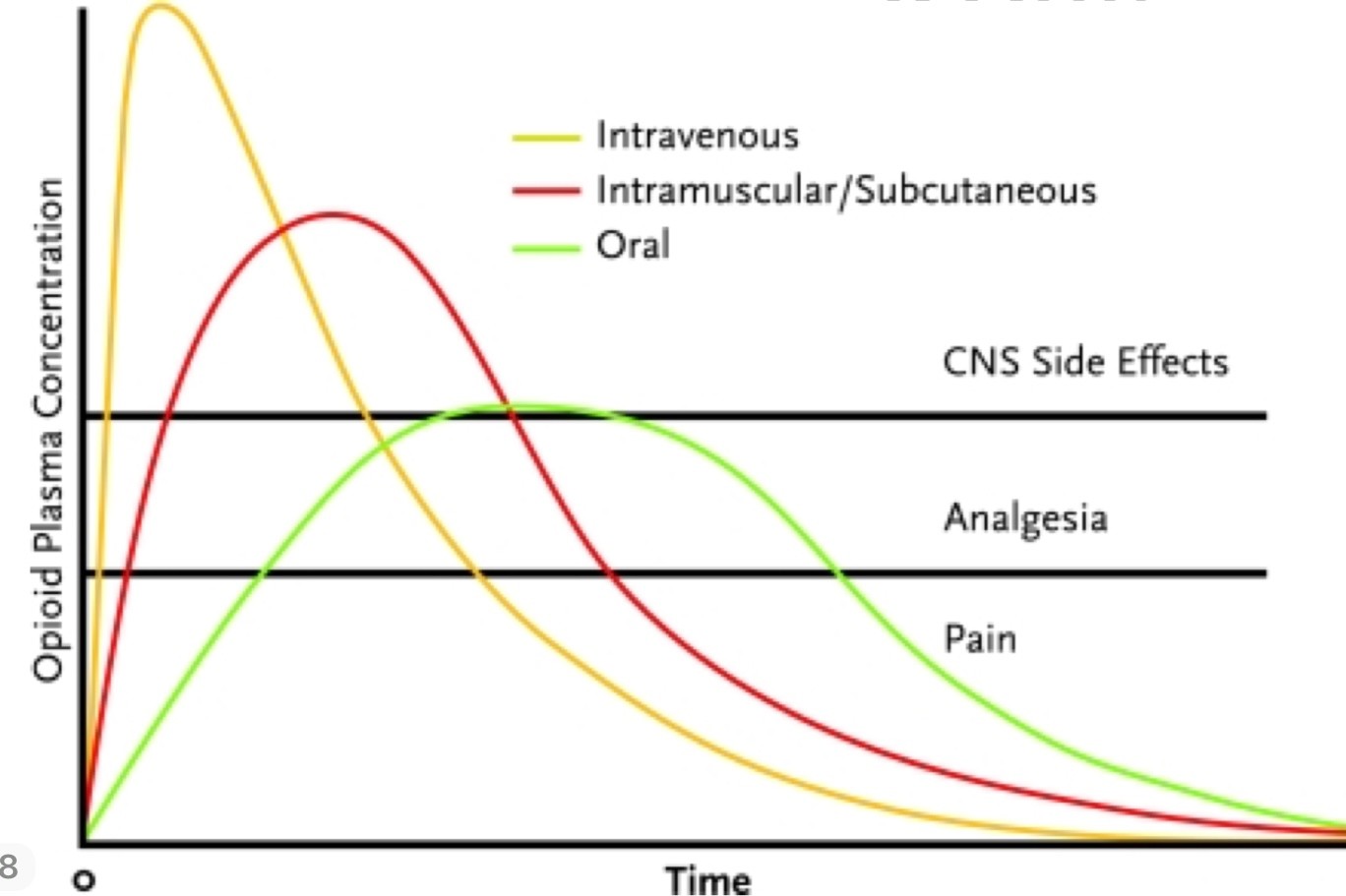
onset (time it takes to get to bottom line)
peak
duration
what is drug half life (T1/2)?
1st order kinetics:time required for drug levels to drop by 50% -> 2 becomes 1 in 2s is 1st half life, 1 becomes 0.5 in 2s is 2nd half life...
0 order kinetics: ratio, drug dose eliminated per unit of time -> 0.5 drinks/hour -> NOT really half life, its constant, doesn't depend on how much drug you take
why do we care about half life?
it determines the dosing schedule
long T1/2: long intervals (ex: 2mg/day)
short T1/2: short intervals (ex: 2mg/3 hours)
is half-life or steady state drug eliminated faster? why?
half-life because most of its elimination happens at beginning
0 kinetic drugs -> proteins responsible for metabolism + excretion are already saturated (working at max capacity)
what is difference w/saturation between 1 and 0 order drugs?
1st order drugs are impossible to saturate in real life impossible to be 0 order (such high concentration would kill patient)
0 order drugs saturate so fast it is impossible to observe 1st order (ex: alcohol)
what is plateau/steady state concentration?
when dose eliminated between 2 administration = dose administered
what is 4 half-life rule?
4 half-lives to reach plateau
4 half-lives to eliminate Rx
how do you manage drug levels?
loading dose: large initial dose to reach peak value fast -> useful for drugs w/long T1/2 or when u want max efficiency rapidly
reduce fluctuations: fixed schedule (even if patient isn't feeling pain at the moment, you know they will), continuous infusion (IV line w/small droplets), depot preparation (large dose w/extended release), dosage + interval reduction (1g 2x/day vs 2g 1x/day)
drug discontinuations: shorter T1/2 is safer, easier to manage toxicity, easier to readjust, exception is detox abuse treatment
how does age affect drug response?
for a same dose, infants + elderly > adults
how does body weight affect drug response?
water:fat ratio affects distribution of drugs
larger individuals require larger dose
ex: for a fat soluble drug, you have to give less to an infant because they have less fat, but need more for water soluble drugs
how does kidney failure affect drug response?
decreased excretion -> increased T1/2
how does liver failure affect drug response?
increases toxicity of drugs or decreases efficacy of pro-drugs
decreases plasma proteins = increases response of drugs with high plasma protein affinity
how does GI transit alterations affect drug response? (ex: diarrhea//constipation)
absorption rate alterations
how does altered electrolytes concentration affect drug response?
ex: dysrhythmias increase is K+ is low
how does comorbidities + polypharmacy affect drug response?
drug interactions if people are taking multiple drugs
how does sex + ethnicity affect drug response?
specific examples
female alcohol metabolism is slower
opioid pain relief efficacy female > male
digoxin increased mortality of female
BiDil only for African Americans
how does diet + patient adherence affect drug response?
healthy diet increases therapeutic benefits
drug-food interactions -> grapefruit juice
decreased plasma proteins due to malnutrition
30-60% patients do not adhere (like follow instructions)
elderly -> memory loss
how does pharmacogenesis affect drug response?
it is the effect of genetic variations on drug responses
some patients metabolize more, some have allergic reactions, some have higher receptor activity
how does tolerance affect drug response?
= decreases response of repeated doses
1. pharmacodynamic tolerance
- changes in receptors
- increases MEC -> need to give higher dose
- may decrease max efficacy
- hysteresis = special short-term dynamic tolerance
2. kinetic tolerance
- # of enzymes working
- usually decrease duration of response
- tachyphylaxis = special short term kinetic tolerance
what is withdrawal/rebound effect?
rebound effect should be opposite of drug effects ex: alcohol is depressant, alcohol withdrawal = muscle tremor/hyperactivity
withdrawal symptom intensity is related to:
- efficacy (proportional)
- dose + duration of intake (proportional)
- half life (inversely proportional)
what is placebo effect?
fraction of drug response based on patient attitudes + expectations of drug effects