Quality Assessment principles
advertisement

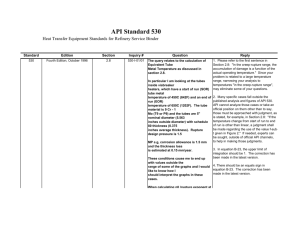
Copenhagen Workshop May 2014 Quality assessment principles Part I L. Paleshnuik Lead Quality Assessor PQP 1 Lynda Paleshnuik | May 2014 Overview Elements of the assessment process Approaches to assessment Using QRM principles Issues of outstanding importance Factors that add complexity Low-importance sections Reviewing the API – 2 common issues The assessment report 2 Lynda Paleshnuik | May 2014 Elements of assessment Assessors must: Review the dossier Write the assessment report (AR) These are equally important aspects. 3 Lynda Paleshnuik | May 2014 Assessment report (AR) Understanding the dossier, the QOS and the AR. The QOS is a template (next slide), filled in by the applicant with a summary of information on the product. The filled QOS becomes the basis for the AR. 4 Lynda Paleshnuik | May 2014 ≈25 pages 5 Lynda Paleshnuik | May 2014 ≈100 pages The assessor reviews the data in the dossier, confirming the QOS is summarized correctly, and adds their comments, questions, discussions. Dossier 6 Lynda Paleshnuik | May 2014 + QOS-PD = AR Brute force method (zero QRM) 7 Lynda Paleshnuik | May 2014 Brute force method In this model the reviewer: Jumps into the review without forethought Reviews every section fully and in depth Assigns equal importance to each section Assigns equal importance to each dossier 8 Lynda Paleshnuik | May 2014 Applying zero QRM 9 Lynda Paleshnuik | May 2014 Don’t read every page of every dossier 10 Lynda Paleshnuik | May 2014 The importance of critical thinking Critical thinking, if it is applied throughout the process, allows for the greatest use of time to result in the highest quality of the review process. Quality risk management is an aspect of critical thinking. Critical analysis is most important when starting a review. 11 Lynda Paleshnuik | May 2014 Understanding quality risk management QRM: the overall and continuing process of managing risks to product quality QRM can be applied to the assessment process (TRS 981 Annex 2, section 5.4, 2013) For quality assessment, major risks include not making the most of limited capacity (excessive resources on lower risk dossiers), and delaying the availability of important products on the market 12 Lynda Paleshnuik | May 2014 Finite capacity 13 Lynda Paleshnuik | May 2014 Decisions during the process 14 Lynda Paleshnuik | May 2014 Brute force method versus using QRM principles You jump into the review without forethought Significant aspects of the dossier are identified (FDC? Solid oral or more complex? Immediate release? Sterile?) You review every section Non-critical, non-important sections are identified You assign equal importance to each section Critical sections are identified to focus attention on 15 Lynda Paleshnuik | May 2014 Making QRM choices The ability to make QRM choices comes from experience. However, there are some basic considerations. 1. Understanding issues of outstanding importance 2. Recognizing factors that add complexity 3. Understanding low-importance sections 16 Lynda Paleshnuik | May 2014 Issues of outstanding importance The dossier and report are in CTD format BUT Some primary issues should be checked first These issues may result in DELAY or TERMINATION of the assessment 17 Lynda Paleshnuik | May 2014 Issues of outstanding importance Primary issues: these may result in termination of the assessment: Issues of the biostudy Biobatch size Correct comparator Acceptable biostudy or biowaiver 18 Lynda Paleshnuik | May 2014 Issues of outstanding importance Primary issues: these may result in termination of the assessment: Issues of the stability studies (without getting into the data): acceptable conditions (“room temperature” vs 30±2ºC, 75±5%), packaging (representing proposed), batches (sufficient number, size). If no evidence of stability or bioavailability, can decide to stop the assessment. Note that a good screening process is helpful here. 20 Lynda Paleshnuik | May 2014 When beginning the review: It is important to understand: 1. the solubility of the API This topic is covered later in this talk Consideration of the API solubility is necessary throughout the assessment process Lynda Paleshnuik | May 2014 When beginning the review: It is important to know: 2.If there are related dossiers that have been reviewed. This is important both for consistency of approach, and to minimize duplication of effort. Lynda Paleshnuik | May 2014 Dossier screening process Examines the completeness of a dossier Checks for key data Only dossiers meeting the minimum standards are accepted for assessment Without a (good) screening process, lower quality dossiers are submitted/assessed, increasing assessment time. One of the largest factors for assessment time is the quality of the dossier. 23 Lynda Paleshnuik | May 2014 Absolute relationship 24 Lynda Paleshnuik | May 2014 Factors that add complexity Unfamiliar or less common dosage form Complex API synthesis or complex dosage form (e.g. inhalation products) FPPs with additional safety issues (e.g. sterile products) Combination products (especially where the APIs are incompatible) Forms with modified release (EC, transdermals, ER) Novel excipients 25 Lynda Paleshnuik | May 2014 Understanding low-importance sections Not all data is of equal importance. When capacity is limited, agencies can decide on areas to spend less time on. (This should be defined, or each assessor will make their own decisions.) General low-importance sections: Batch packaging records (most solid orals) Compendial excipient specifications and COAs Validation of GC methods for common solvents 26 Lynda Paleshnuik | May 2014 Understanding low-importance sections Examples of agency-specific low-importance sections: Less/no assessment of validation of methods for companies with a good track record No review of batch records when there is a strong inspectorate The decision is based on the capacities of the NMRA, including laboratories, assessors and inspectors 27 Lynda Paleshnuik | May 2014 Examples approaches to method validation Applicant is new to stringent requirements: review their validation data in detail; check their calculations for precision, linearity, etc; review their chromatograms, review SST Applicant is not new but errors have been identified in the past: review their validation summary, spotcheck raw data, review chromatograms, review SST Applicant is established: review their validation summary, review impurity/assay chromatograms for separation, review SST 28 Lynda Paleshnuik | May 2014 System suitability testing Unlike validation, SST is part of the running of the method. Regardless of the approach to assessment of method validation, SST should always be reviewed as part of the method review for purity, assay, dissolution, residual solvent methods. This is important due to the inherent variability of columns, column aging, variation in MP and eluting solvent, instrumentation, etc. 29 Lynda Paleshnuik | May 2014 SST criteria – non-compendial methods HPLC assay: precision (RSD ≤ 1% (API) or ≤ 2% (FPP)) and: either peak asymmetry/tailing factor (≤ 2) or theoretical plates (≥ 2000) or resolution (≥ 2) HPLC/GC purity: precision (RSD ≤ 5%*, up to 10% at LOQ) and resolution (≥ 2) TLC purity: limit of detection and specificity (API/FPP/placebo spiked with impurities at their specified limits) *30 May consider up to 10% for 0.2-0.5% level Lynda Paleshnuik | May 2014 API – applicants must… Applicants must demonstrate: They have a valid source of the API They have adequate understanding of the API (physicochemical properties, impurities, etc) Common issue #1 They can adequately control the quality of the API (specifications, validated methods, reference standards) Common issue #2 The API is sufficiently stable 34 Lynda Paleshnuik | May 2014 Elements of API sections Applicants must: Provide data on the API according to one of the 4 options (PQ’d API, CEP, APIMF, full data). Regardless of the option used, the dossier should include sufficient info to show their understanding of the API (solubility profile and hygroscopicity are critical, as well as the impurity profile) The elements that must be demonstrated by the applicant, must be understood and discussed by the assessor in the AR. 35 Lynda Paleshnuik | May 2014 Factors that add complexity: API API is a product of fermentation – an understanding of fermentation processes is required API is sterile – understanding of sterilization processes is required API is low solubility – common issue with additional considerations (covered next) API is hygroscopic or moisture sensitive – considerations for manufacture (e.g. avoid wet granulation) and packaging to ensure stability; one standard test = BP = EP 5.11 36 Lynda Paleshnuik | May 2014 Low solubility API The solubility of the API over the physiological pH should be determined for every API The dose solubility volume (DSV) should be calculated: DSV = largest dosage strength (mg) Minimum concentration of API (mg/mL)* DSV > 250 mL is a low solubility API * Lowest solubility determined over pH 1.2-6.8 at 37◦C 37 Lynda Paleshnuik | May 2014 Low solubility API Polymorphism must be investigated and controlled if necessary, and particle size is critical. These parameters are critical for any API with DSV > 250 mL, i.e. any low solubility API. An exception can be made when the manufacture of the FPP involves fully dissolving the API. This is common with reproductive health products, where an organic solvent is often used for this purpose. In this case neither PSD nor polymorphism is relevant. 38 Lynda Paleshnuik | May 2014 Low solubility API Identifying critical issues: Eg. Dossier where polymorphism is critical (BCS low solubility, different polymorphs observed). Recent report correctly states it is critical, but does not deal with it. This is not meaningful unless you understand the implications/necessary considerations and include them in the report. Polymorphism: are there various forms? is one preferred? Is there a test in specs? Does it ensure that form (e.g. XRD, DSC, IR)? What was the form of the API lot used in the BE study? Confirm the reference standard has the same form. Ensure the specs include the validated test/limits (validated to show it can distinguish possible forms). 38 Lynda Paleshnuik | May 2014 API Solubility categories BCS high solubility: the API soluble over the entire physiological pH range (DSV < 250 mL) BCS low solubility: The DSV is calculated based on the lowest solubility over the physiological pH range. The API is often still soluble at other pH(s) in that range. (DSV > 250 mL) Critically low solubility: The API is insoluble over the entire phys pH range. (DSV > 250 mL) An example follows regarding the change in an API supplier. 39 Lynda Paleshnuik | May 2014 PSD and solubility There are three situations: API is BCS high solubility i.e. soluble over the physiological pH range PSD is not critical API is BCS low solubility (DSV > 250 mL) but is soluble at some phys pH PSD is critical, a test/limits must be in API specifications (based on BE profile) API is critically low soluble (API is insoluble over the physiological pH range) PSD is critical, a test/limits must be in API specifications (based on BE profile) PSD should be in the retest parameters 45 Lynda Paleshnuik | May 2014 PSD and solubility API is critically low soluble (API is insoluble over the physiological pH range) PSD is critical, a test/limits must be in API specifications (based on BE profile) PSD should be in the retest parameters Note that PSD should be carefully considered, case-by-case. Normal PSD limits are: d10 NMT x d50 xx-xxx d90 NMT xxxx For critically low soluble, consider the need for a range for d90. 45 Lynda Paleshnuik | May 2014 Low solubility API QRM approach to low solubility APIs The API has been found to have low solubility What should be checked next (far ahead in the CTD dossier), before getting into PSD and polymorphism? Is it fully dissolved during manufacture. If so, you have saved time reviewing PSD/polymorphism data (no longer important considerations). (in 21 QOS subsections) 40 Lynda Paleshnuik | May 2014 Common API Issues Multiple suppliers or change in supplier FPP manufacturer’s use of API supplier’s methods 41 Lynda Paleshnuik | May 2014 Common API issue #1 – multiple API suppliers or change in API supplier It is acceptable to have more than one supplier of an API as long as the quality of the API is adequately controlled for each supplier. When it is a low solubility API, the PSD and polymorphic form (if relevant) should be representative of the lot(s) used in the BE study, for API from each supplier. 41 Lynda Paleshnuik | May 2014 Change in API supplier A common FPP manufacturer complaint: they have conducted the BE study with API from supplier A, but that supplier is no longer available (or will not provide sufficient info on the API). Can they change the API supplier? 42 Can they change the supplier without having to repeat the BE studies using the new source? Lynda Paleshnuik | May 2014 Change in API supplier Full information is required on API from the new supplier, AND The quality of the API must be adequately controlled for each supplier. This does not mean they must have the same impurity profile, but they must have adequate controls based on their impurity profile (according to potential impurities, residual solvents, etc) When it is a low solubility API, the PSD and polymorphic form (if relevant) should be representative of the lot(s) used in the BE study, for API from each supplier. (See details for PSD, next slides) If the above is established, new BE studies are not required and no new stability data is required on the FPP, beyond the commitment for studying of future batches 43 Lynda Paleshnuik | May 2014 Change in API supplier PSD The generally accepted variations from BE lot PSD results are: D10 ± 45% D50 ± 30% D90 ± 45% Where the values are < 10 μm, the values are doubled. 44 Lynda Paleshnuik | May 2014 Change in API supplier PSD change There are three situations: API is soluble over the physiological pH range PSD is not critical API is low solubility (DSV > 250 mL) but is soluble at some phys pH PSD is critical, a change must be within the tolerances, OR Comparative dissolution may support differences outside these tolerances API is critically low soluble (API is insoluble over the physiological pH range) PSD is critical, a change must be within the tolerances Change outside these tolerances may require a new biostudy 45 Lynda Paleshnuik | May 2014 PSD change supported by dissolution data For the low solubility API, where it is soluble at some phys pH: Comparative dissolution profiles can be used to justify a change in PSD outside the tolerances: Multipoint dissolution profiles in 3 media (pH 1.2 or 0.1N HCl, pH 4.5 and 6.8) without surfactant for: One FPP batch made with the API with the new PSD profile One FPP batch made with the API with PSD within the tolerances of the BE lot Dissolution profile data on the BE lot 46 Lynda Paleshnuik | May 2014 Common API issue #2 The applicant’s use of the API supplier’s methodology The applicant must be able to adequately control the quality of the API Often the applicant adopts the supplier’s methods How much verification and/or assessment is required for the validation? There are three basic scenarios, and in each case we are assuming we have the full validation from the originating site 47 Lynda Paleshnuik | May 2014 The applicant’s use of the API methodology: Validation required 1) When analytical methods have been developed in a GMP compliant facility and the original site included intermediate precision, pursuit of method transfer data is not required. 2) When both sites are within the same corporation, method transfer data isn't usually required. If there is a concern, the applicant should confirm that a transfer report is available. 48 Lynda Paleshnuik | May 2014 The applicant’s use of the API methodology: Validation required 3) For methods that come from a facility for which GMP compliance hasn't been established (e.g. APIMF holder), the applicant is required to provide: • complete validation, or, • method transfer (specificity and intermediate precision), or, 49 Lynda Paleshnuik | May 2014 The applicant’s use of the API methodology: Validation required Equivalency: for Results of NLT 2 batches at each test site. For assay methods: e.g. COAs by both sites the same two batches. For impurity methods, results (both sites) for samples spiked with the specified impurities at their specification limits. 50 Lynda Paleshnuik | May 2014 Final assessment report (AR) The AR is a very important document. The quality of the AR is one of the biggest factors of the success of the assessment process. That AR will be used throughout the rounds of assessment of the dossier, and beyond: In the review of variations As a reference document for other dossiers A good AR is useful as a training tool 51 Lynda Paleshnuik | May 2014 Absolute relationship 52 Lynda Paleshnuik | May 2014 Data we need in the first assessment report/round Characterization of API lot used in the BE study specially PSD/polymorphism of low solubility API FPP dissolution data (biolot) in media over the physiological pH range Discriminatory data for dissolution method when required (CR products, critically low soluble API) This data should be requested as soon as possible. Lynda Paleshnuik | May 2014 Final assessment report (AR) Wrapping up the report will be covered in the second talk on quality assessment principles. 54 Lynda Paleshnuik | May 2014 Summary Elements of the assessment process Approaches to assessment Using QRM principles Issues of outstanding importance Factors that add complexity Low-importance sections Reviewing the API – 2 common issues The assessment report 55 Lynda Paleshnuik | May 2014 56 Lynda Paleshnuik | May 2014