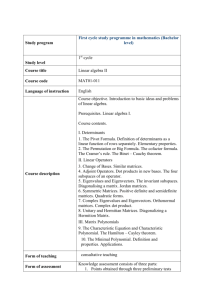
Computational Biology, Part 7
Similarity Functions and
Sequence Comparison with Dot
Matrices
Robert F. Murphy
Copyright 1996, 1999-2001.
All rights reserved.
Similarity Functions
Used to facilitate comparison of two
sequence elements
logical valued (true or false, 1 or 0)
test
whether first argument matches (or could
match) second argument
numerical valued
test
degree to which first argument matches
second
Logical valued similarity
functions
Let Search(I)=‘A’ and Sequence(J)=‘R’
A Function to Test for Exact Match
MatchExact(Search(I),Sequence(J))
would return
FALSE since A is not R
A Function to Test for Possibility of a Match
using IUB codes for Incompletely Specified
Bases
MatchWild(Search(I),Sequence(J))
since R can be either A or G
would return TRUE
Numerical valued similarity
functions
return value could be probability (for DNA)
Let
Search(I) = 'A' and Sequence(J) = 'R'
SimilarNuc (Search(I),Sequence(J)) could return 0.5
since chances are 1 out of 2 that a purine is adenine
return value could be similarity (for protein)
Let
Seq1(I) = 'K' (lysine) and Seq2(J) = 'R' (arginine)
SimilarProt(Seq1(I),Seq2(J)) could return 0.8
since lysine is similar to arginine
usually use integer values for efficiency
Scoring (similarity) matrices
For each pair of characters in alphabet,
value is proportional to degree of similarity
(or other scoring criterion) between them
For proteins, most frequently used is
Mutation Data Matrix from Dayhoff, 1978
(MDM78)
Dayhoff PAM250 similarity
matrix (partial)
A
B
C
D
E
F
G
H
A
2
0
-2
0
0
-4
1
-1
B
0
0
-4
3
2
-5
0
1
C
-2
-4
12
-5
-5
-4
-3
-3
D
0
3
-5
4
3
-6
1
1
E
0
2
-5
3
4
-5
0
1
F
-4
-5
-4
-6
-5
9
-5
-2
G
1
0
-3
1
0
-5
5
-2
H
-1
1
-3
1
1
-2
-2
6
Origin of PAM 250 matrix
Take aligned set of closely related proteins
For each position in the set, find the most common
amino acid observed there
Calculate the frequency with which each other
amino acid is observed at that position
Combine frequencies from all positions to give
table showing frequencies for each amino acid
changing to each other amino acid
Take logarithm and normalize for frequency of
each amino acid
Sequence comparison with dot
matrices
Goal: Graphically display regions of
similarity between two sequences (e.g.,
domains in common between two proteins
of suspected similar function)
Sequence comparison with dot
matrices
Basic Method: For two sequences of
lengths M and N, lay out an M by N grid
(matrix) with one sequence across the top
and one sequence down the left side. For
each position in the grid, compare the
sequence elements at the top (column) and
to the left (row). If and only if they are the
same, place a dot at that position.
Sequence comparison with dot
matrices - References
W.M. Fitch. An improved method of testing
for evolutionary homology. J. Mol. Biol.
16:9-16 (1966)
W.M. Fitch. Locating gaps in amino acid
sequences to optimize the homology
between two proteins. Biochem. Genet.
3:99-108 (1969)
Sequence comparison with dot
matrices - References
A.J. Gibbs & G.A. McIntyre. The diagram,
a method for comparing sequences. Its use
with amino acid and nucleotide sequences.
Eur. J. Biochem. 16:1-11 (1970)
A.D. McLachlan. Test for comparing related
amino acid sequences: cytochrome c and
cytochrome c551. J. Mol. Biol. 61:409-424
(1971)
Sequence comparison with dot
matrices - References
J. Pustell & F.C. Kafatos. A high speed, high
capacity homology matrix: zooming
through SV40 and polyoma. Nucleic Acids
Res. 10:4765-4782 (1982)
J. Pustell & F.C. Kafatos. A convenient and
adaptable package of computer programs
for DNA and protein sequence management,
analysis and homology determination.
Nucleic Acids Res. 12:643-655 (1984)
Examples for protein sequences
(Demonstration A5, Sequence 1 vs. 2)
(Demonstration A5, Sequence 2 vs. 3)
Interpretation of dot matrices
Regions of similarity appear as diagonal
runs of dots
Reverse diagonals (perpendicular to
diagonal) indicate inversions
Reverse diagonals crossing diagonals (Xs)
indicate palindromes
(Demonstration A5,
Sequence 4 vs. 4)
Interpretation of dot matrices
Can link or "join" separate diagonals to
form alignment with "gaps"
Each
a.a. or base can only be used once
Can't
trace vertically or horizontally
Can't double back
A gap
is introduced by each vertical or
horizontal skip
Uses for dot matrices
Can use dot matrices to align two proteins
or two nucleic acid sequences
Can use to find amino acid repeats within a
protein by comparing a protein sequence to
itself
Repeats
appear as a set of diagonal runs stacked
vertically and/or horizontally
(Demonstration A5,
Sequence 5 vs. 6)
Uses for dot matrices
Can use to find self base-pairing of an RNA
(e.g., tRNA) by comparing a sequence to
itself complemented and reversed
Excellent approach for finding sequence
transpositions
Filtering to remove “noise”
A problem with dot matrices for long
sequences is that they can be very noisy due
to lots of insignificant matches (i.e., one A)
Solution use a window and a threshold
compare
character by character within a
window (have to choose window size)
require certain fraction of matches within
window in order to display it with a “dot”
Example spreadsheet with
window
(Demonstration A6)
How do we choose a window
size?
Window size changes with goal of analysis
size
of average exon
size of average protein structural element
size of gene promoter
size of enzyme active site
How do we choose a threshold
value?
Threshold based on statistics
using
shuffled actual sequence
find
average (m) and s.d. () of match scores of
shuffled sequence
convert original (unshuffled) scores (x) to Z scores
• Z = (x - m)/
use
using
threshold Z of of 3 to 6
analysis of other sets of sequences
provides
“objective” standard of significance
Displaying matrices by Pustell
method with MacVector
Goal: Determine differences in
arrangements of elements of pBluescript
family of vectors
Starting point: Use sequences of three of the
members of the family: open the first three
files in the Common Vectors: Bluescript
folder.
Dot matrices with MacVector
From Analyze menu select Pustell DNA matrix. Dialog appears.
Dot matrices with MacVector
Select SYNBL2KSM and SYNBL2SKM. Use defaults for all else.
Dot matrices with MacVector
23 reagons of homology (“diagonals”) obtained. Request “Matrix
map” only (don’t need “Aligned sequences”)
Dot matrices with MacVector
Note inversion near nucleotide 700 (the direction of the polylinker is
reversed between the two vectors)
Dot matrices with MacVector
To examine effect of threshold, decrease “min. % score” from 65 to 55
Dot matrices with MacVector
Now we get many (223) diagonals.
Dot matrices with MacVector
Note presence of many short regions of at least 55% homology.
Dot matrices with MacVector
Now increase threshold to 90%.
Dot matrices with MacVector
Now just 3 diagonals are found.
Dot matrices with MacVector
Note absence of short homologous regions (“noise”).
Dot matrices with MacVector
Now compare SYNBL2KSP to SYNBL2SKM.
Dot matrices with MacVector
22 diagonals found using default settings.
Dot matrices with MacVector
Note second large inversion at one end of sequences.
More dot matrices with
MacVector - DNA homology
Goal: Duplicate Figure 6 of Chapter 3 of
Sequence Analysis Primer
Get Accession numbers J02289 (Polyoma)
and J02400 (SV40) from Entrez
Do Pustell DNA Matrix analysis using
parameters similar to those used in text
(window size = 41, %identity = 51)
More dot matrices with
MacVector - DNA homology
More dot matrices with
MacVector - DNA homology
More dot matrices with
MacVector - DNA homology
More dot matrices with
MacVector - protein homology
Goal: Reproduce Figure 15 from Chapter 3
of Sequence Analysis Primer
Get Accession numbers P17678 (Chicken)
and X17254 (human) erythroid transcription
factors using Entrez
Do Pustell Protein Matrix Analysis
Reading for next class
B & O, Chapter 7 just pp. 145-155
Additional optional reading: Sequence
Analysis Primer, pp. 124-134 “Dynamic
Programming Methods” (on web site as
Reading 1)
(03-510) Durbin et al, Sections 2.1 - 2.4
Everybody: Look over paper by Needleman
and Wunsch on web site (Reading 2)
Summary, Part 7
Similarity functions or similarity matrices
describe (quantitatively) the degree of
similarity between two sequence elements
(bases or amino acids)
The Dayhoff MDM78 matrix is a similarity
matrix commonly used to estimate the
degree to which a change from one amino
acid to another can be “tolerated” in a
protein
Summary, Part 7
Dot matrices graphically present regions of
identity or similarity between two sequences
The use of windows and thresholds can
reduce “noise” in dot matrices
Inversions, duplications and palindromes
have unique “signatures” in dot matrices