Molecular dynamics
advertisement

Molecular Dynamics
Molecular dynamics
Some random notes on molecular dynamics simulations
Seminar based on work by Bert de Groot and many
anonymous Googelable colleagues
Molecular Dynamics
Most material in this seminar has been produced by Bert de Groot at the
MPI in Göttingen.
Molecular Dynamics
Molecular Dynamics
Schrödinger equation
Born-Oppenheimer approximation
Nucleic motion described classically
Empirical force field
Molecular Dynamics
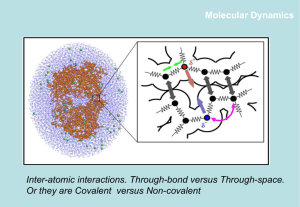
Inter-atomic interactions
Molecular Dynamics
d2
() ma 2 R Eel ( R1 ,..., RN ), 1,..., N .
dt
Motions of nuclei are described classically:
Non-bonded interactions
Covalent bonds
Eibond
approximated
exact
=
=
R
KBT {
0
|R|
Potential function Eel describes the electronic influence on motions of the nuclei and is
approximated empirically „classical MD“:
E el
Bindungen
i
E ibond
E
Bindungs
winkel j
angle
j
E
Dihedral
winkel k
dihe
k
.
rep .
vdW
( ECoul
E
E
,
,
, ) ...,
„ForceField“
Molecular Dynamics
Possible ‘extras’:
Planarity
Hydrogenbond
Weird metal
Induced charge
Multi-body interaction
Pi-Pi stacking
and a few more
Non-bonded interactions
Lennard-Jones potential
Molecular Dynamics
Coulomb potential
Molecular Dynamics
Molecular Dynamics
Now we need to give all atoms some initial speed, and then, evolve that speed over
time using the forces we now know. The average speed of nitrogen in air of 300K is
about 520 m/s. The ensemble of speeds is best described by a Maxwell distribution.
Back of the enveloppe calculation:
500 m/s = 5.10 12Å/s
Let’s assume that we can have things fly 0.1 A in a
straight line before we calculate forces again, then we
need to recalculate forces every 20 femtosecond
(one femtosecond is 10 -15sec.
In practice 1 fsec integration steps are being used.
http://en.wikipedia.org/wiki/Verlet_integration
http://en.wikipedia.org/wiki/Maxwell_speed_distribution
Molecular Dynamics
Knowing the forces (and some randomized
Maxwell distributed initial velocities) we can
evolve the forces over time and get a trajectory.
Simple Euler integration won’t work as this
figure explains. And as the rabbit knows...
You can imagine that if you know where you
came from, you can over-compensate a bit.
These overcompensation algorithms are called
Verlet-algorithm, or Leapfrog algorithm.
If you take bigger time steps you overshoot your goal. The Shake algorithm can fix that.
Shake allows you larger time steps at the cost of little imperfection so that longer
simulations can be made in the same (CPU) time.
http://en.wikipedia.org/wiki/Verlet_integration
Molecular Dynamics
Molecule: (classical) N-particle system
d2
m i 2 ri Fi ( r )
dt
Newtonian equations of motion:
Fi ( r ) iV ( r )
r ( r1 ,..., rN )
Integrate numerically via the „leapfrog“ scheme:
with
Δt 1fs!
(equivalent to the Verlet algorithm)
Molecular Dynamics
Solve the Newtonian equations of motion:
Molecular Dynamics
Molecular dynamics is very expensive ...
Example: A one nanosecond Molecular Dynamics simulation of F1ATPase in water (total 183 674 atoms) needs 106 integration steps, which
boils down to 8.4 * 1017 floating point operations.
on a 100 Mflop/s workstation:
ca 250 years
...but performance has been improved by use of:
+ multiple time stepping
ca. 25 years
+ structure adapted multipole methods*
ca.
6 years
+ FAMUSAMM*
ca.
2 years
+ parallel computers
ca. 55 days
* Whatever that is
Molecular Dynamics
Molecular Dynamics
Role of environment - solvent
Explicit or implicit?
Box or droplet?
Molecular Dynamics
periodic boundary conditions
Molecular Dynamics
Molecular Dynamics
Limits of MD-Simulations
classical description:
chemical reactions not described
poor description of H-atoms (proton-transfer)
poor description of low-T (quantum) effects
simplified electrostatic model
simplified force field
incomplete force field
only small systems accessible (104 ... 106 atoms)
only short time spans accessible (ps ... μs)
Molecular Dynamics
H. Frauenfelder et al., Science 229 (1985) 337
Molecular Dynamics
Molecular Dynamics
One example: Thermodynamic Cycle
A
D
B
C
A -> B -> C -> D -> A
ΔG=0!
Molecular Dynamics
At Radboud you have seen in
‘Werkcollege 3 Thermodynamica’:
Folded 105 C
?
1
Unfolded 105 C
3
Folded 75 C
Unfolded 75 C
2
And, for Radboud students only, I type here the answer in Dutch…
ΔT kan natuurlijk in Celcius of Kelvin) en is dan of 0 of 105-75=30
Cp is heat capacity en kan temepartuuronafhankelijk verondersteld
worden. Cp(unfolded)-Cp(folded)=6.28 kJ/molK.
Proces 1 is isobaar dus dH1=Cp(folded)*dT
Proces 3 is isobaar dus dH3=Cp(unfolded)*dT
Proces 2 is isotherm dus ΔH2=ΔH(unfolding;75 C)=509kJ/mol
Vul alle getallen in en je krijgt ΔH(unfolding; 105 C)=697.4 kJ/mol.
Molecular Dynamics
Thermodynamic Cycle in bioinformatics
ΔG1
A
B
ΔG2
ΔG4
D
C
ΔG3
ΔG1+ΔG2+ΔG3+ΔG4=0 =>
ΔG1+ΔG3=-ΔG2-ΔG4
So if you know the difference
between ΔG2 and ΔG4, you also
know the difference between
ΔG1 and ΔG3 (and vice versa).
Obviously, all arrows should be bidirectional equilibrium-arrows, but if I draw them that
way we are sure to start getting the signs wrong. …
Molecular Dynamics
The relations between energy, force and time can
be simulated in MD. Obviously you cannot simply
put a force on an atom for some time and
calculate the Energy from the force, path, and
time.
But for now, we forget all calibrations, etc, and
end up with Energy = Force * time
Molecular Dynamics
Stability of a protein is ΔG-folding, which is the
ΔG of the process Protein-U <-> Protein-F
ΔG(fold)wt
Wt-U
Wt-F
ΔG(mut)F
ΔG(mut)U
Mut-U
ΔG(fold)mut
Mut-F
So we want ΔG(fold)wtΔG(fold)mut; which is
impossible.
But we can calculate
ΔG(mut)F-ΔG(mut)U;
which gives the same
number!
Molecular Dynamics
Such cycles can be set up for ligand binding, for
membrane insertion, for catalysis, etc.
Don’t be surprised if you have to work out a
similar cycle in the exam…