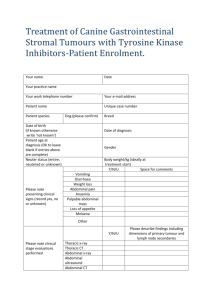
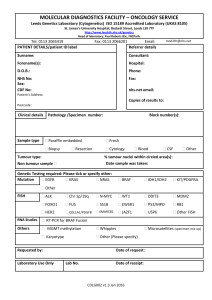
Appendix 13: LOREC Histopathology Dissection and Reporting
advertisement

Appendix 1: Acute Radiation morbidity Scoring (o) (1) (2) (3) (4) Skin No change over baseline Folicular, faint erythema/epilation/dry decreased sweating or dull desquamation/ Tender or bright erythema, patchy moist desquamation/moderate edema Confluent, moist desquamation other than skin fold, pitting edema Ulceration, hemorrhage, necrosis Mucous membrane No change over bas Injection/may experience mild pain not requiring analgesic Patchy mucositis which may produce an inflammatory serosanguinitis discharge/may experience moderate pain requiring analgesia Confluent fibrinous/mucositis/may include severe pain requiring narcotic Ulceration, hemorrhage or necrosis Upper GI No Anorexia with <=5% weight loss from pretreatment baseline/nausea not requiring antiemetics/abdominal discomfort not requiring parasympatholytic drugs or analgescis Anorexia with <=15% weight loss from pretreatment baseline/nausea &/or vomiting requiring antimetics/ abdominal pain requiring analgesics Anoredia with >15% weight loss from pretreatment baselineor requiring N-G tube or parenteral support. Nausea &/or vomiting requiring tube or parenteral support/abdominal pain, severse despite medication/hematemesis or melena/abdominal distention (flat plate radiograph demonstrates distended bowel loops) Ileus, subacute or acute obstruction performation, GI bleeding requiring transfustion/abdominal pain requiring tube decompression or bowel diversion change Lower GI including pelvis No change Increased frequency or change in quality of bowel habits not requiring medication/rectal discomfort not requiring analgesics Diarrhea requiring parasympatholytic drugs (e.g., lomotil)/mucous discharge not necessitating sanitary pads/ rectal or abdominal pain requiring analgesics Diarrhea requiring parenteral support severe mucous or blood discharge necessitating sanitary pags/abdominal distention (flat plate radiograph demonstrates distended bowel loops) Acute or subacute obstruction, fistula or perforation; GI bleeding requiring transfusion; abdominal pain or tenesmus requiring tube decompression or bowel diversion Genitourinar y No change Frequency of urination or nocturia twice pretreatment habit/ dysuria, urgency not requiring medication Frequency of urination or nocturia which is less frequent than every hour. Dysuria, urgency bladder spasm requiring local anesthetic (e.g., pyridium) Frequency with urgency and nocturia hourly or more frequently / dysuria, pelvis pain or bladder spasm requiring regular, frequent narcotic/gross hematuria with/without clot passage Hematuria requiring transfusion/acute bladder obstruction not secondary to clot passage, ulceration or necrosis Hematologic WBC (X 1000) >=4.0 3.0 - < 4.0 2.0 - < 3.0 1.0 - <2.0 <1.0 Platelets (X 1000) >=100 75 - <100 50 - <75 25 - <50 <25 or bleeding Neutrophils >=1.9 1.5 - <1.9 1.0 - <1.5 0.5 - < 1.0 < 0.5 or sepsis Hemoglobin (GM %) >11 11-9.5 < 9.5 – 7.5 < 7.5 – 5.0 ----- spontaneous Guidelines: The acute morbidity criteria are used to score/grade toxicity from radiation therapy. The criteria are relevant from day 1, the commencement of therapy, through day 90. Thereafter, the EORTC/RTOG criteria of late effects are to be utilized. The evaluator must attempt to discriminate between disease – and treatment – related signs and symptoms. An accurate baseline evaluation prior to commencement of therapy is necessary. All toxicities Grade 3, 4 or 5* must be verified by the Principal Investigator. *ANY TOXICITY WHICH CAUSED DEATH IS GRADED 5. Appendix 2: Dindo Grade Definition for recording of post-operative complications Grade I- Any deviation from the normal postoperative course without the need for pharmacological treatment or surgical, endoscopic, and radiological interventions Allowed therapeutic regimens are: drugs as antiemetics, antipyretics, analgetics, diuretics, electrolytes, and physiotherapy. This grade also includes wound infections opened at the bedside Grade II- Requiring pharmacological treatment with drugs other than such allowed for grade I complications. Blood transfusions and total parenteral nutrition are also included Grade III- Requiring surgical, endoscopic or radiological intervention Grade IIIa- Intervention not under general anesthesia Grade IIIb- Intervention under general anesthesia Grade IV- Life-threatening complication (including CNS complications)* requiring IC/ICU management Grade IVa- Single organ dysfunction (including dialysis) Grade IVb- Multiorgan dysfunction Grade V- Death of a patient Suffix “d” If the patient suffers from a complication at the time of discharge (see examples in Table 2), the suffix “d” (for “disability”) is added to the respective grade of complication. This label indicates the need for a follow-up to fully evaluate the complication. *Brain hemorrhage, ischemic stroke, subarrachnoidal bleeding, but excluding transient ischemic attacks. Appendix 3: Dworak tumor regression grading system: TRG0- No regression TRG 1- predominantly tumor with significant fibrosis/ vasculopathy TRG 2- predominantly fibrosis with scattered tumor cells, easy to find TRG 3- only scattered tumor cells, difficult to find, in the space of fibrosis with/without acellular mucin TRG 4- no viable tumor cells detectable Appendix 4: INNOVA Randomization notepad Part A- Identifying details Patient’s full name:……………………………………………………….. Gender: M / F Age (years): Hospital ID:………………………… Part B- Radiological assessment 1. Predominant tumor site in rectum? Lower third □ 2. Radiological stage: T-stage: N-stage: 3. Least Distance from outer edge of tumor/ metastatic node to mesorectal fascia(mm): Middle third□ M-stage: Upper third□ Part C- Eligibility checklist (If shaded boxes are ticked, patient not eligible for INNOVA) 4. Histologically confirmed adenocarcinoma? Yes□ No□ 5. Age 18 to 70 years? Yes□ No□ 6. ECOG performance status 0-2? Yes□ No□ 7. At least one of the following present: Yes□ No□ Resectable cT4 disease or T3 tumor in the lower rectum / T3c or T3d in mid rectum/ Involved or threatened MRF (tumor or node extends to ≤2mm of the mesorectal fascia)/ N2 disease/ Extramural vascular invasion 8. Any metastatic lateral pelvic nodes? Yes□ No□ 9. Any established distant metastasis? Yes □ No□ 10. Adequate haematological function? Yes□ No□ 11. Adequate liver function? Yes □ No□ 12. Adequate renal function? Yes □ No□ 13. Any active stable or unstable angina, Yes□ No□ NYHA class 2 congestive cardiac failure, myocardial infarction in the past 1 year, acute coronary syndrome even if controlled, arrhythmias? 14. Does patient have other serious medical comorbidity? Yes □ No□ 15. Any synchronous or prior cancer in past 5 years Yes□ No□ 16. Any prior radiotherapy to pelvis? Yes □ No□ 17. Any previous treatment for current cancer? Yes □ No□ 18. If female patient-pregnant or lactating? Yes□ No□ 19. Known HIV, HBV or HCV infection? Yes□ No□ 20. Has the patient given informed consent? Yes □ No□ Is the patient eligible for randomization into INNOVA? Yes □ No □ If patient is eligible, but will not be randomized, reason? Patient preference □ Other reason (specify) □ Investigator preference □ Part D- Randomization- Treatment allocation Randomized to: Arm A-neoadjuvant chemotherapy INNOVA ID: □ Arm B-interval chemotherapy □□□ Physician name: ………………………………………………. Signature: ………………………… Date of randomization (dd/mm/yyyy)………………………………………… □ Appendix 5: MRI Imaging Sequences The protocol uses a turbo spin-echo T2-weighted technique with a surface pelvic phasedarray coil, with 3-mm sections and a small field of view. The images obtained have an inplane resolution of 0.6 x 0.6 mm. For all tumors, imaging is performed perpendicular to the long axis of the tumor. Coronal imaging is performed for all tumors arising at or below the levator muscle origins. Images are stored in DICOM format on compact disks. In each patient, extramural depth of tumor invasion is measured, with the electronic calipers of the workstation, as the maximum depth of penetration beyond the outer edge of the longitudinal muscle layer. The closest distance between tumor and the mesorectal fascia is recorded. Potential circumferential resection margin involvement by tumor is defined as tumor, tumor deposit, or involved lymph node abutting or extending through the mesorectal fascia or extending to less than or equal to 2 mm from the mesorectal fascia. No bowel preparation, air insufflation, or intravenous antispasmodic agents are used. Intravenous contrast enhancement or intrarectal contrast agents such as water or gel are not used. With a 1.5-T sigma HD MR imager (GE Healthcare), the following sequences are used: FOV Slice thickness Spacing Frequency x Phase NEX AX 2D FIESTA SAG T2 Ax SSFSE BH AXIAL T2 COR T2 AXIAL T1 FSE DWI B 1000 AXIAL T1 BH 40 26 32 24 20 26 31 32 8.0 3.0 5.0 3.0 3.0 5.0 5.0 5.0 1.0 1.0 1.0 0.5 0.5 1.0 1.5 1.0 224x288 320x256 224x192 256x224 256x224 320x192 128x128 320x192 1.0 4.0 0.55 4.0 4.0 3.0 1.0 1.0 Appendix 6: INNOVA Pre-randomization radiological assessment Part A- Identifying details Patient name:……………………………………………….………… Age (years):................................ Hospital ID:………………………… Date of examination:...................................... Date of reporting:…………………… Radiologist:..................................................... …… Signature…………………………… Part B- Tumour Location Predominant tumor site in the rectum? - □ Middle 1/3 (>6 to ≤12 cm from anal verge) □ Lower 1/3 (≤6cm from anal verge) □ Mid and lower third □ Upper 1/3 (>12cm from anal verge) Is tumour peritonealised or non-peritonealised? (strike off whichever is not applicable) Is the levator involved? Yes/No Is the intersphincteric groove involved? Yes/No Part C- Radiological Staging - Length (cm)…………………………. - Wall thickness (mm)………………… Maximum distance of tumour beyond Muscularis propria (mm)................. Maximum tumour Local Invasion: Submucosa T1 □ Muscularis propriaT2 □ T3c □ Peritoneal T4 □ Beyond Muscularis propria: T3a □ □ □ T3b T3d Adjacent organs T4 Is definite extra mural vascular invasion present? Yes / No No of perirectal lymph nodes seen: Lateral pelvic nodes? Present/ Absent No. metastatic nodes (short axis ≥15 mm) : Distance of largest node/tumour from nearest mesorectal fascial margin (mm): MRF threatened : Yes / No Longest dimension of pathological node (short axis) – Node 1 Node 2 No. of nodes with diffusion restriction: N stage M stage □N0 □M0 □N1 (1-3 nodes) □ N2 (4+Nodes) □M1 (specify site)…………………………….. Clinical stage: cT N M Eligible for Innova? (see eligibility criteria below) : Yes □ No □ (Involved or threatened MRF (tumor or node extends to ≤2mm of or beyond the MRF), Any T3 tumor in the lower rectum,T3c or T3d in mid rectum (ie-tumor extending >5mm beyond the muscularis propria), Resectable T4, T3/T4 N1 or T1-T4 N2, Extramural vascular invasion) □ Appendix 7A: INNOVA Participant Information Sheet- English Cancer Institute (WIA) Name of Principal Investigator: Dr.Ramakrishnan A S Tel.No(s). 044-22350231/ 9840085569 Title of study: INNOVA: Interval or Neoadjuvant chemotherapy in rectal cancer. A phase II randomized study comparing interval versus neoadjuvant chemotherapy in magnetic resonance imaging-defined high risk rectal cancer Why are we doing this study? Surgery is the main treatment for rectal cancer. However, in many patients, radiation and chemotherapy is also required. In such patients who require all three modalities of treatment, the standard practice nowadays is to first give radiation with chemotherapy for 5 to 6 weeks and then do the surgery after another 6 to 8 weeks. Once the patient recovers from the surgery, he/she will be given chemotherapy for another 5 to 6 months. Radiation and surgery is useful in treating the cancer in the rectum and nearby nodes. In patients with advanced rectal cancers, there is a high chance of having undetectable disease in other parts of the body and hence, chemotherapy is used to treat undetectable disease in other parts of the body. Although the advances in surgery and radiation techniques have considerably reduced the chances of the disease coming back near the operated area, the disease may come back in other parts of the body in many patients in spite of getting chemotherapy. One possible reason for this could be that in the currently followed treatment schedule, adequate dose of chemotherapy is given only after radiation and surgery, ie- nearly 4 months after starting the treatment. This delay in giving chemotherapy may cause the undetectable disease in other areas of the body to grow further. Hence, it may be a good idea to give chemotherapy at an earlier stage in the treatment schedule. That is what we are planning to do in this study. We want to see if giving at least half of the chemotherapy before surgery will lead to a better response of the tumor, which in turn will predict a better outcome. We are therefore comparing two new treatment schedules and if we find that one of them is better than the other, we are planning to compare that treatment with the standard treatment at a later date. There are some encouraging reports of giving chemotherapy at an earlier time from studies conducted in foreign countries. How will we do the study? In this study conducted at the Cancer Institute (WIA), Chennai, once we confirm that you are eligible to participate in this study, we will explain the details of this study to you. Once you give the informed consent to take part in the study after fully understanding about the study, then you will be treated by one of the two treatment schedules of the study. In Arm A, you will first receive 3 cycles of chemotherapy (injection and tablet) for 9 weeks. Then you will receive radiation and a chemotherapy tablet for 6 weeks. After another 6 weeks, surgery will be performed. Following surgery, after 4-8 weeks, you will again receive 3 more cycles of chemotherapy. In Arm B, you will first receive radiation and a chemotherapy tablet for 6 weeks. After 4 weeks, you will receive 3 cycles of chemotherapy (injection and tablet) for 9 weeks. You will then undergo surgery and 4 to 8 weeks later, you will receive 3 more cycles of chemotherapy. -----------------------------(Signature/ Left thumb impression of participant) Date: Place: The decision regarding which type of treatment you will undergo will not be taken by you or the doctor treating you. Instead, you will be assigned to one of the two treatment schedules by a computer program. This is the standard practice in these types of studies to ensure that the study is not influenced by anyone and the results are genuine. This study will include 47 patients in each of the two groups and will be done in 2 stages. In the first stage, we will include 21 patients in each arm. If we find a big difference in the tumor response between the two arms, then we will stop the study. If we do not find a big difference in the first stage, then we will include 26 more patients in each arm. After your surgery, the tumor which has been removed will be sent for biopsy and will be studied in detail to know how the tumor has responded to the treatment. Photographs of the tumor will also be taken and stored. However, all your treatment and personal details will be kept confidential. All complications during the treatment will be recorded. The results of this study will be again analyzed after all 94 patients have completed treatment. The duration of treatment will be the same regardless of which type of surgery you undergo. You will also receive all three modalities of treatment for rectal cancer- radiation, surgery and chemotherapy. The only difference will be the type of treatment you will receive firstchemotherapy or radiation. In this study, surgery will be performed at a later time when compared to the standard treatment. Some similar studies done in other places have not reported any serious complications due to either of the two treatment regimens. The risks and complications are expected to be similar between the two types of treatments. Progression of the rectal cancer during treatment has not been a problem in the earlier studies. The doctors involved in this study (including surgery, radiation and chemotherapy doctors) have a lot of experience in treating rectal cancer patients. If you choose to join this study, you will be doing so entirely on your own free will without any coercion from anyone. Also, you are free to withdraw from the study at any time after you have given your consent. While you may or may not have any benefit in joining the study, if the results of the study show that the tumor responds more with one type of treatment, these patients could possibly have a better outcome. Also, the doctors may have better information to decide which type of treatment benefits patients who come in the future. The cost of treatment will depend on your category decided by the hospital, but will be the same regardless of which type of treatment you undergo. You will be expected to bear the entire cost of the treatment according to your category. In case of any doubts, you can contact your doctor at the above mentioned telephone number. In case of any adverse events encountered in this study, you can contact the principal investigator or the ethical committee for compensation. The need for/ amount of compensation will be decided by the ethical committee as empowered by the Government of India gazette notification. -----------------------------(Signature/ Left thumb impression of participant) Date: Place: This is to certify that a copy of this information sheet has been given to the patient and that the contents have been explained to the patient in a language that he understands in my presence. (I also certify that the translation to the vernacular is accurate to the best of my knowledge) ------------------------------ Signatures of the Principal Investigator Date: Place: I hereby declare that I have been given a copy of this information sheet and that I have read it carefully/ the contents have been explained to me in details by the doctor in a language that I understand --------------------------------------------(Signatures / Left Thumb Impression) Date: Place: Name of the Participant: ____________________________________ Son / Daughter / Spouse of:__________________________________ Complete postal address: _____________________________________ Appendix 7C: INNOVA Participant Information sheet (Telugu) `క్యాన్సర్ ఇన్స్టిట్యాట్ (W.I.A.) పయల్గొనే వయళ్ళూ అధ్ాయన్ం ట్ట్ ై ల్ ి : సంప్రదంచండి: మల క్యాన్సర్ విరయమం లేదా న్యా అన్ుబంధ క్ీమోథెరపీ. మాగ్నెటిక్ రనసో నాన్సస ఇమేజంగ్ నిరవచంచబడిన్ ప్రమాదం మల క్యాన్సర్ న్యా అన్ుబంధ క్ీమోథెరపీ వరనసస్ విరయమం పో లచడం దశ యాదృచిక అధాయన్ం. ఎందుకు మేము ఈ అధాయన్ం చేసు ున్ె: సు చ ర క్ిత్స ప్రధాన్ అయితే అనేక రోగులలో మల క్యాన్సర్ చక్ిత్స, రేడియిేషన్స మరియు రసయయన్ కూడా అవసరం ఉంద. చక్ిత్స మూడు ప్దధ త్ులన్ు అవసరం ఎవరు అలాంటి రోగులకు, పయరమాణిక ప్దధ తి రోజులోో మొదటి 5 6 వయరయల రసయయన్ విక్ిరణానిె ఇవయవలని మరియు త్రువయత్ మరొక 6 న్ుండి 8 వయరయల త్రయవత్ శసు చ ర క్ిత్స చేయడానిక్ి ఉంద. రోగ్ి దేర్ శసు చ ర క్ిత్స న్ుండి తిరిగ్ి ఒకసయరి అత్న్ు / ఆమె మరొక 5 6 నెలల రసయయన్ ఇవవబడుత్ుంద. రేడియిేషన్స మరియు శసు చ ర క్ిత్స ప్ురీషనాళం మరియు ఆధునిక మల క్యన్సర్ రోగుల ఏ నోడససమీప్ంలో క్యన్సర్ చక్ిత్సలో ఉప్యోగప్డుత్ుంద, శరీరం యొకక ఇత్ర పయరంతాలోో వీలుప్డదు వయాధ కలిగ్ి మరియు అందుక్ే, క్నమోథెరపీ లో వీలుప్డదు వయాధ చక్ిత్సకు ఉప్యోగ్ిసు యరు ఎకుకవ అవక్యశం ఉంద అందుక్ే శరీరం మరియు ఇత్ర పయరంతాలోో, క్నమోథెరపీ శరీర ఇత్ర పయరంతాలోో వీలుప్డదు వయాధ చక్ిత్సకు ఉప్యోగ్ిసు యరు. శసు చ ర క్ిత్స మరియు రేడియిేషన్స టెక్ిెక్స అభివృదధ గణనీయంగ్య అమలు పయరంత్ం సమీప్ంలో తిరిగ్ి వచేచ వయాధ అవక్యశయలు త్గ్ిిన్ అయితే, వయాధ భయమే ఉన్ెప్పటిక్ీ తిరిగ్ి అనేక రోగులు లో శరీరం యొకక ఇత్ర పయరంతాలోో రయవచుచ. ఈ ప్రసు ుత్ం త్రువయత్ చక్ిత్స షెడయాల్, క్ీమోథెరపీ త్గ్ిన్ మోతాదు లో భావించాలిస క్ోసం ఒక క్యరణం అంటే దాదాప్ుగ్య 4 నెలల చక్ిత్స పయరరంభమెైన్ త్రయవత్, మాత్రమే రేడియిేషన్స మరియు శసు చ ర క్ిత్స త్రయవత్ ఇవవబడుత్ుంద. క్ీమోథెరపీ ఇవవడం లో క్యలయాప్న్ మరింత్ పెరుగుతాయి శరీరంలోని ఇత్ర పయరంతాలోో వీలుప్డదు వయాధ క్యరణం క్యవచుచ, అందుక్ే, ఇద చక్ిత్స షెడయాల్ లో లోపిస్తు ప్రమాదమే రసయయన్ ఇవవడం మంచ ఆలోచన్ క్యవచుచ. మేము ఈ అధాయన్ంలో చేయండి ప్రణాళిక్య ఏమి ఉంద. మేము శసు చ ర క్ిత్స ముందు క్ీమోథెరపీ కనీసం సగం ఇవవడం, కరమంగ్య ఒక మంచ ఫలితానిె అంచనా ఇద కణితి, ఒక మంచ త్ప్పకుండా ఉంటాయి చయడాలన్ుకుంటే. అందువలన్ మేము రనండు క్ొత్ు చక్ిత్స షెడయాల్ పో లచడం ఉంటాయి మరియు మేము వయటిని ఒక ఇత్ర కంటే మెరుగ్నైన్ అని అన్ుకుంటే, మేము ఆ తేదన్ పయరమాణిక చక్ిత్స తో చక్ిత్స పో లచడానిక్ి చేసు ునాెరు. విదేశీ దేశయలలో నిరవహంచన్ అధాయనాలు న్ుండి ఒక ముందు సమయంలో రసయయన్ ఇవవడం క్ొనిె పో ర త్సహంచడం నివేదకలు ఉనాెయి. మేము అధాయన్ం ఎలా చేసు ుంద? మీరు ఈ అధాయన్ంలో పయలగినేందుకు అరహత్ అని నిరయధరించండి ఒకసయరి క్యాన్సర్ ఇనిటిటయాట్ (WIA) చెనెనె వదద నిరవహంచన్ ఈ అధాయన్ంలో, మేము మీరు ఈ అధాయన్ం యొకక వివరయలన్ు వివరిసు ుంద మీరు. మీరు అధాయన్ం గురించ ప్ూరిుగ్య అవగ్యహన్ త్రువయత్ అధాయన్ం పయలగిన్డానిక్ి ఔషధానిె ఇచేచ అప్ుపడు మీరు అధాయన్ం రనండు చక్ిత్స ప్రణాళిక దావరయ చక్ిత్స సయధాం. చేయి ఒక లో మీరు మొదటి 9 వయరయలు రసయయన్ యొకక 3 చక్యరల (ఇంజనక్షన్స మరియు టాబలో ట్) అందుకుంటారు. అప్ుపడు మీరు 6 వయరయలు రేడియిేషన్స మరియు ఒక రసయయన్ టాబలో ట్ అందుకుంటారు. మరో 6 వయరయల శసు చ ర క్ిత్స ఉంట ంద త్రయవత్. శసు చ ర క్ిత్స త్రువయత్, 4-8 వయరయల త్రయవత్ మీరు మళ్ళీ మీరు 6 వయరయలు రేడియిేషన్స మరియు రసయయన్ టాబలో ట్ అందుకుంటారు చేయి బి క్ీమోథెరపీ 3 ఎకుకవ సయరుో అందుకుంటారు. 4 వయరయల త్రయవత్, మీరు 9 వయరయలు రసయయన్ యొకక 3 చక్యరల (ఇంజనక్షన్స మరియు టాబలో ట్) అందుకుంటారు. మీరు శసు చ ర క్ిత్స మరియు 4 న్ుండి 8 వయరయల త్రువయత్, మీరు రసయయన్ యొకక 3 మరినిె చక్యరల అందుకుంటారు. (పయలగినేయొకకసంత్కం/ఎడమబొ టన్వేలిముదర) తేదీ సథ లము సథ లము చక్ిత్స ఏ రకం సంబంధంచన్ నిరణయం మీరు ప్రయాణంలో క్ింర ద మీరు తీసుకున్ె క్యదు ఉంట ంద లేదా డాకటర్ చక్ిత్స. బదులుగ్య, మీరు ఒక కంప్ూాటర్ పో ర గ్యరమ్ దావరయ రనండు చక్ిత్స ప్రణాళిక క్ేటాయించబడుత్ుంద. ఈ అధాయన్ం ఎవరనైనా ప్రభావిత్ం క్యదు నిరయధరించడానిక్ి అధాయనాలు ఈ రకమెైన్ పయరమాణిక ఆచరణ మరియు ఫలితాలు నిజమెైన్ ఉంటాయి. ఈ అధాయన్ం రనండు సమూహాలలో ప్రతి 47 రోగులు ఉనాెయి మరియు 2 దశలోో ప్ూరిు అవుత్ుంద. మొదటి దశలో మేము ప్రతి చేతి లో 21 రోగులు కలిగ్ి ఉంట ంద. మేము రనండు చేత్ులు మధా కణితి ప్రతిసపందన్ పెదద తేడా కన్ుగ్ొనేందుకు మేము కణితి పెదద తేడా లేకపో తే, అప్ుపడు మేము అధాయన్ం చేయవు. ఈ అధాయన్ం 47 రోగులకు రనండు సమూహాలలో ప్రతి కలిగ్ి ఉంట ంద మరియు 2 దశలోో ప్ూరిు అవుత్ుంద. మొదటి దశలో. మేము రనండు చేత్ులు మధా కణితి ప్రతిసపందన్ పెదద తేడా కన్ుగ్ొనేందుకు మేము అప్ుపడు మేము అధాయన్ం చేయవు, ప్రతి చేతి 21 రోగులు కలిగ్ి ఉంట ంద. మేము మొదటి దశలో పెదద తేడా లేకపో తే. అప్ుపడు మేము ప్రతి చేతి లో 26 మరింత్ రోగులు కలిగ్ి ఉంట ంద. మీ శసు చ ర క్ిత్స త్రయవత్, తొలగ్ించబడింద ఇద కణితి బయాపీస క్ోసం ప్ంప్బడుత్ుంద మరియు కణితి చక్ిత్స సపందచారు ఎలా వివరయలు అధాయనాలు ఉంట ంద. కణితి ఛాయాచతారలన్ు కూడా తీసుకున్ె మరియు నిలవ చేయబడుత్ుంద. అయితే, అనిె మీ చక్ిత్స మరియు వాక్ిుగత్ మెగ్యవయటో అణు ఉంట ంద. చక్ిత్స సమయంలో అనిె సమసాలు న్మోదు చేయబడుత్ుంద. అనిె 94 రోగులు చక్ిత్స ప్ూరు యిాన్త్రువయత్ ఈ అధాయన్ం యొకక ఫలితాలు మళ్ళీ విశలోషించవచుచ ఉంట ంద. చక్ిత్స వావధ సంబంధం లేకుండా మీరు గురిక్యవలస్ి శసు చ ర క్ిత్స ఏ రకం ఒక్ే ఉంట ంద. మీరు కూడా మాత్రమే తేడా మీరు మొదటి క్నమోథెరపీ లేదా రేడియిేషన్స అందుకుంటారు చక్ిత్స రకం ఉంట ంద మల క్యాన్సర్ రేడియిేషన్స, శసు చ ర క్ిత్స మరియు రసయయన్ చక్ిత్స మూడు ప్దధ త్ులన్ు అందుకుంటారు. పయరమాణిక చక్ిత్స పో లిస్తు ఈ అధాయన్ంలో త్రయవత్ చేయబడుత్ుంద. ఇత్ర ప్రదేశయలలో జరుగుత్ుంద క్ొనిె ప్రిశోధన్లు రనండింటి వెనదా నియమాలు గ్యని క్యరణంగ్య ఎలాంటి సమసాలు లేవని. ప్రమాదం మరియు సమసాలు చక్ిత్సలు రనండు రక్యల మధా ఉండాలని భావిసుునాెరు. చక్ిత్స సమయంలో మల క్యాన్సర్ గమన్ం ముందు అధాయనాలోో ఒక సమసా క్యదు. (శసు చ ర క్ిత్స, రేడియిేషన్స మరియు రసయయన్ వెనదుాలు సహా) ఈ అధాయన్ంలో పయలగిన్ె వెనదుాలు అన్ుభవం మల క్యాన్సర్ రోగులు చక్ిత్స చాలా. మీరు ఈ అధాయన్ంలో చేరడానిక్ి ఎంచుకుంటే, మీరు ఎవరనైనా న్ుండి ఏ నిరబంధానిె లేకుండా మీ స్తవచచన్ు క్యబటిట ప్ూరిుగ్య చేయడం. కూడా మీరు మీ ప్తాకశీరిికలక్నక్యకయి త్రయవత్ సమయంలో అధాయన్ం ఉప్సంహరించుక్ోవడంతో ఉచత్ం. కణితి చక్ిత్స ఒకటి రకంతో మరింత్ సపందంచ ఇందులో ఫలితాలు, ఈ రోగులకు బహుశయ మెరుగ్నైన్ ఫలిత్ం కలిగ్ి ఉంటే మీరు లేదా, అధాయన్ం చేరడానిక్ి ఎలాంటి ప్రయోజన్ం పో వచుచ అయితే. కూడా, వెనదుాలు భవిషాత్ు ు లో వచచన్ చక్ిత్స ప్రయోజనాలు రోగుల ఇద రకం నిరణయించుకుంటారు మంచ సమాచారయనిె కలిగ్ి ఉండవచుచ చక్ిత్స ఖరుచ ఆసుప్తిర నిరణయించుకుంద మీ వరి ం ఆధారప్డి ఉంట ంద, క్యనీ సంబంధం లేకుండా మీరు గురిక్యవలస్ి చక్ిత్స ఏ రకం ఒక్ే ఉంట ంద. మీరు మీ వరి ం ప్రక్యరం చక్ిత్స మొత్ు ం ఖరుచ భరించలేక అవసరం లేదు. అన్ుమానాలన్ు సంభవించన్ ప్క్షంలో, మీరు పెనన్ పతరొకన్ె టెలిఫో న్స న్ంబర్ మీ డాకటర్ సంప్రదంచవచుచ. భారత్దేశం గ్నజట్ నోటిఫిక్ేషన్స ప్రభుత్వం అధక్యరం ఈ అధాయన్ంలో ఎదురొకంద ఏ ప్రతికూల ఈవెంట్స సందరభంలో, మీరు ప్రధాన్ ప్రిశోధకుడిని లేదా నెనతిక కమిటీ సంప్రదంచవచుచ. (పయలగినేయొకకసంత్కం/ఎడమబొ టన్వేలిముదర) తేదీ సథ లము ఈ సమాచారం ప్త్రం క్యపీని రోగ్ిక్ి మరియు విషయాలు ద నా సమక్షంలో అరథం ఒక భాష థా రోగ్ిక్ి వివరించారు చేస్ిన్ ఇవవబడింద అని సరిటఫెన ఉంద. (నేన్ు కూడా పయరంతీయ అన్ువయద ఖచచత్మెైన్ అని సరిటఫెన ) నా విజఞాన్ ఉత్ు మ పయలగినేయొకకసంత్కం/ఎడమబొ టన్వేలిముదర) తేదీ సథ లము నేన్ు దీనిె ఇందుమూలముగ్య నేన్ు ఈ సమాచారం ప్త్రం క్యపీని ఇవవడం జరిగ్ింద మరియు నేన్ు జఞగరత్ుగ్య చదవయలని / విషయాలు నేన్ు అరథం భాషలో వెనదుాడు వివరయలు నాకు వివరించారు ప్రకటిసు ుంద. పయలగినేయొకకసంత్కం/ఎడమబొ టన్వేలిముదర) తేదీ సథ లము పయలగినే యొకక పతరు _________________________ కుమారుడు / కుమారను / భరు _________________________ ప్ూరిు చరునామా __________________________ Appendix 8A:INNOVA Participant Informed Consent Form- English Cancer Institute (WIA) Participant identification number for this trial: _______________________ Title of project: INNOVA: Interval or Neoadjuvant chemotherapy in rectal cancer. A phase II randomized study comparing interval versus neoadjuvant chemotherapy in magnetic resonance imaging-defined high risk rectal cancer Name of Principal Investigator: Dr.Ramakrishnan A S Tel.No(s). 044-22350231/ 9840085569 The contents of the information sheet dated that was provided have been read carefully by me / explained in detail to me, in a language that I comprehend, and I have fully understood the contents. I confirm that I have had the opportunity to ask questions. The nature and purpose of the study and its potential risks / benefits and expected duration of the study, and other relevant details of the study have been explained to me in detail. I understand that my participation is voluntary and that I am free to withdraw at any time, without giving any reason, without my medical care or legal right being affected. I understand that the information collected about me from my participation in this research and sections of any of my medical notes may be looked at by responsible individuals from Cancer Institute (WIA). I give permission for these individuals to have access to my records. I agree to take part in the above study. --------------------------------------------(Signatures / Left Thumb Impression) Date: Name of the Participant: ____________________________________ Son / Daughter / Spouse of:__________________________________ Complete postal address: _____________________________________ Place: This is to certify that the above consent has been obtained in my presence. -----------------------------Signatures of the Principal Investigator 1) Witness – 1 Date: Place: 2) Witness – 2 -----------------------------Signatures -------------------------------Signatures Name: Address: Name: Address: NB Three copies should be made, for (1) patient, (2) researcher, (2) Institution Appendix 8C: INNOVA Participant Informed consent form (Telugu) పయలగినే సమాచారం అంగ్ీక్యరప్త్రంపెన (PICF) క్యాన్సర్ ఇనిటిటయాట్(WIA) ఈ విచారణ క్ోసం పయలగినే గురిుంప్ు సంఖా; ______________ పయరజనకటు శీరిిక: INNOVA: మల క్యాన్సర్ విరయమం లేదా న్యా అన్ుబంధ క్ీమోథెరపీ. రనండో దశ యాదృచిక మాగ్నెటిక్ రనసో నాన్సస ఇమేజంగ్ నిరవచంచబడిన్ ప్రమాదం మల క్యాన్సర్ న్యా అన్ుబంధ క్ీమోథెరపీ వరనసస్ విరయమం పో లచడం అధాయన్ం. దేర్ ప్రధాన్ ప్రిశోధకుడిని పతరు: DR రయమకృషణ న్స వంటి Tel.No(s) 044-22350231/9840085569 అందంచంద ______ నాటి వయళళీ విషయాలు నాకు జఞగరత్ుగ్య చదవండి చేశయరు /నేన్ు గరహంచడానిక్ి భాషలో, నాకు వివరంగ్య వివరించారు, మరియు నేన్ు ప్ూరిుగ్య ఆయన్నే చేశయరు. నేన్ు ప్రశెలు అడగండి అవక్యశం నిరయధరించండి. అధాయన్ం మరియు దాని సంభావా ప్రమాదాల సవభావం మరియు ప్రయోజన్ం / అబ్దద అధాయన్ం వావధ అంచనా లాభాలన్ు ఆరిిసు ుంద మరియు అధాయన్ం ఇత్ర సంబంధత్ వివరయలు వివరంగ్య నాకు వివరించాడు చేశయరు. నేన్ు రక్షణ లేదా చటట ప్రమెైన్ హకుక ప్రభావిత్మెైన్ అరథం. నా పయలగిన్డం సవచింద మరియు నేన్ు ప్రభావిత్మెైన్ నా వెనదా సంరక్షణ లేదా చటట ప్రమెైన్ హకుక లేకుండా, ఏ క్యరణం లేకుండా, ఏ సమయంలో ఉప్సంహరించుక్ోవయలని ఉచత్. నేన్ు ఈ ప్రిశోధన్ మరియు నా వెనదా గమనికలు ఏ విభాగ్యలలో నా పయలగినేటటో నాకు గురించ స్తకరించన్ సమాచారయనిె ఈ వాకుులు నా రిక్యరుులు యాక్నసస్ క్ోసం (WIA) నేన్ు అన్ుమతి ఇవయవలని క్యాన్సర్ ఇనిటిటయాట్ బాధాత్ వాకుులు చయశయరు కలుగవచచని. నేన్ు పెనన్ అధాయన్ం పయలగిన్డానిక్ి అంగ్ీకరిసు ునాెరు పయలగినేయొకకసంత్కం/ఎడమబొ టన్వేలిముదర) తేదీ సథ లము పయలగినే యొకక పతరు :__________________________ కుమారుడు / కుమారను / భరు :_____________________ ప్ూరిు చరునామా ______________________________ ఈ పెనన్ అన్ుమతి నా సమక్షంలో ప ందన్ అని సరిటఫెన ఉంద ______________________________ ప్రధాన్ ప్రిశోధ్కుల్ల్ో సంతకం తేదీ సథ ల్ము 1. సయక్షి 1 2.సయక్షి -2 సంత్క్యలు సంత్క్యలు సంత్క్యలు సంత్క్యలు సంత్క్యలు సంత్క్యలు NB:మూడు క్యపీలు 1 క్ోసం చేయవలస్ిన్వి. రోగ్ి 2. ప్రిశోధకుడు 3. సంసథ Appendix 9: CAPOX schedule and dose modifications for INNOVA The following applies for all cycles of CAPOX regimen, whether pre or post-operative, neoadjuvant or interval. The chemotherapy treatment is given in three-weekly schedules as follows: Treatment schedule (21 day cycle): Day 1, 0:00 IV bolus palensetron 0.25mg (or equivalent) IV bolus dexamethasone 8 mg flush line with 5% dextrose 0.00 – 2:00 oxaliplatin 130 mg/m2 IV infusion, 2 hrs, 250 ml 5% dextrose 2:00 flush line with 5% dextrose Day 1, evening capecitabine 1000 mg/m2 p.o. Day 2-14 capecitabine 1000 mg/m2 p.o. twice daily Day 15, morning capecitabine 1000 mg/m2 p.o. Day 16-21 no treatment Notes: For patients who are obese (BMI >30 approx), the surface area used to calculate doses of oxaliplatin and capecitabine should be capped using the following scheme: Patient‟s height (cm): Cap surface <150 150-159 160-169 170-179 180+ 1.5 1.7 1.9 2.1 2.3 area (m2) at: Notes: The treatment cycle includes 28 capecitabine doses taken 12-hourly. This starts with the evening dose on day 1 and ends with the morning dose on day 15. The capecitabine dose is rounded to the nearest achievable dose. Patients are instructed to take capecitabine within 30 minutes after food, approximately 12 hourly (e.g. 8 am and 8pm). Because of a potential in vitro chemical reaction between oxaliplatin and chloride ions, care is taken to avoid contact with normal saline in the drip tubing etc. Do not use injection equipment containing aluminium. Oral antiemetics (starting day 2): Dexamethasone 4 mg tds x1 day; 4 mg bd x1 day; 4 mg od x1 day. Domperidone or metoclopramide prn Note on the use of dexamethasone: For patients at high risk of steroid side effects (e.g. diabetics) or for those who develop toxicity attributable to steroids (e.g. dyspepsia; dysphoria; etc), the oral steroid should be omitted and “p.r.n.” oral 5HT3 inhibitor given. Scheduled tests FBC and clinical assessment (NCI toxicity scores) should be performed on the day of starting each cycle, (or within 3 working days before) and the results available before starting. Biochemistry (including creatinine, bilirubin, and either AST or ALT) is done at the same time as FBC; these results should be available before starting the cycle. Toxicity and dose adjustments for OxCap Haematological -Check CBC on (or up to 3 working days before) day 1 of each cycle. Delay 1 week if neutrophils < 1.5 x 109/l or platelets < 75 x 109/l. Only treat when neutrophils and platelets are above these limits. -If >1 delay, or 1 delay of 2 weeks occurs, reduce the capecitabine and oxaliplatin doses by 20% and continue at the lower dose for subsequent cycles unless further toxicity occurs. -If a further delay(s) for myelotoxicity occurs despite a 20% reduction, a further dose reduction may be made, at the discretion of the treating investigator. Neurotoxicity -Oxaliplatin commonly causes transient paraesthesia of hands, feet and sometimes throat, precipitated by cold and lasting up to a few days after each dose. This does not require treatment or dose reduction. -With cumulative dosing, some patients develop more severe neurotoxicity, requiring omission of oxaliplatin. For example: o persistent paraesthesia occurring in warm as well as cold conditions with significant discomfort o Numbness with loss of function (eg dropping objects) or pain Renal function - Before starting, ensure patient fulfils eligibility for renal function, i.e. normal blood urea and serum creatinine. -Thereafter, if serum creatinine rises above normal limit, and >25% from baseline, check EDTA clearance. A significant deterioration in renal function should be investigated to exclude a postoperative complication or disease progression/relapse. Hepatobiliary function -Bilirubin 1.25 x ULN and transaminase (either AST or ALT) 3 x ULN is required for study entry. -Patients on capecitabine may have temporary treatment-related elevation of transaminases, which require interruption of treatment. Other alterations in hepatic function should be investigated to exclude a postoperative complication or disease progression/relapse. -If, after investigation, treatment is to continue, the following adjustments should be made: . AST/ALT >5x ULN: withhold chemotherapy until recovered below this limit . Bilirubin >3x ULN: reduce both capecitabine and oxaliplatin by 50% Stomatitis and Diarrhoea -Patients should be provided with routine mouthcare. Loperamide should be provided for symptomatic treatment of diarrhoea -Grade 1 toxicity is managed symptomatically and does not usually require dose reduction or interruption -For any toxicity of grade 2 or higher, stop capecitabine and treat symptomatically until the toxicity has resolved to grade 0 or 1. o NB: when capecitabine is stopped for toxicity the doses are omitted, not delayed. If resolution to grade 0–1 occurs before day 14, capecitabine is resumed for the remainder of the planned cycle; otherwise wait until the next cycle. -When resuming after a pause for toxicity, use the following dose reduction scheme: o Grade 2 toxicity: resume at the same dose after first pause, but reduce both capecitabine and oxaliplatin to 80% of the previous doses if a second pause is required. o Grade 3 toxicity: resume at 80% of original doses (both capecitabine and oxaliplatin) o Grade 4 toxicity: discontinue permanently. -If further toxicity of grade 2 occurs after a dose-reduction, the doses should be reduced by a further 20%. Hand-foot syndrome (HFS) -Treat symptomatically. Pyridoxine 50 mg tds by mouth or topical corticosteroid may help. -If HFS is still a problem, interrupt capecitabine and treat symptomatically until the toxicity has resolved to grade 0 or 1, then resume with a 20% dose reduction. DPD deficiency; cardiotoxicity -With any fluoropyrimidine regimen, the occasional patient is encountered (approx 1-3%) who has markedly exaggerated toxicity due to reduced catabolism. If this occurs, await full recovery. Further treatment at much reduced capecitabine dose (e.g. 50%) or with single agent oxaliplatin may be considered. Please discuss with the CI or one of the clinical co-investigators. -Capecitabine may provoke angina attacks or even MI in patients with ischaemic heart disease. In this event the risks of continuing adjuvant treatment are likely to out-weigh the benefits, and discontinuation is recommended. Hypomagnesaemia Management -Patients should be evaluated and managed as per local medical practice. If hypomagnesaemia is present, replacement should be managed as per local medical practice. A patient‟s serum magnesium level should be at or above 0.41 mmol/L throughout the study. It is important to assess and manage serum potassium and ionized calcium (corrected for albumin) in patients who have hypocalcaemia. Allergic reactions to oxaliplatin -Approx. 0.5% patients develop acute hypersensitivity to oxaliplatin, usually after more than 6 cycles. During drug administration, the patient may develop rash, fever, swollen mouth or tongue, hypo- or hypertension and other signs/symptoms of hypersensitivity. This rarely develops to fullblown anaphylaxis, even with repeated treatment -If acute hypersensitivity occurs, discontinue the infusion and treat with IV corticosteroid and antihistamine -After full recovery, the patient may continue with that cycle‟s capecitabine. -At the investigator‟s discretion, the patient may be rechallenged with oxaliplatin at the next cycle. In this case, premedication is recommended as follows: Dexamethasone 4mg p.o. 6 hourly starting 24 hours pre-treatment, + 8mg IV 30 minutes pre-dose Chlorphenamine 10mg (or equivalent) + ranitidine 50mg (or equivalent) IV 30 minutes pre-dose. Continue dexamethasone, chlorphenamine and ranitidine for 24-48 hours after treatment with oxaliplatin. Appendix 10: RECIST criteria v1.1 Assessment of overall tumour burden and measurable disease To assess objective response or future progression, it is necessary to estimate the overall tumour burden at baseline and use this as a comparator for subsequent measurements. Measurable disease is defined by the presence of at least one measurable lesion. At baseline, tumour lesions/lymph nodes will be categorised measurable or non-measurable as follows: Measurable Tumour lesions: Must be accurately measured in at least one dimension (longest diameter in the plane of measurement is to be recorded) with a minimum size of: • 10mm by CT scan (CT scan slice thickness no greater than 5 mm) • 10mm caliper measurement by clinical exam (lesions which cannot be accurately measured with calipers should be recorded as non-measurable). • 20mm by chest X-ray. Malignant lymph nodes: To be considered pathologically enlarged and measurable, a lymph node must be ≥15mm in short axis. At baseline and in follow-up, only the short axis will be measured and followed Non-measurable All other lesions, including small lesions (longest diameter <10mm or pathological lymph nodes with ≥10 to <15mm short axis) as well as truly non-measurable lesions. Lesions considered truly non-measurable include: leptomeningeal disease, ascites, pleural or pericardial effusion, inflammatory breast disease, lymphangitic involvement of skin or lung, abdominal masses/ abdominal organomegaly identified by physical exam that is not measurable by reproducible imaging techniques. Measurement of lesions All measurements should be recorded in metric notation, using calipers if clinically assessed. All baseline evaluations should be performed as close as possible to the treatment start and never more than 4 weeks before the beginning of the treatment. Method of assessment The same method of assessment and the same technique should be used to characterise each identified and reported lesion at baseline and during follow-up. Imaging based evaluation should always be done rather than clinical examination unless the lesion(s) being followed cannot be imaged but are assessable by clinical exam. Chest X-ray: Chest CT is preferred over chest X-ray, particularly when progression is an important endpoint, since CT is more sensitive than X-ray, particularly in identifying new lesions. However, lesions on chest X-ray may be considered measurable if they are clearly defined and surrounded by aerated lung. CT, MRI: CT is the best currently available and reproducible method to measure lesions selected for response assessment. MRI is also acceptable in certain situations (e.g. for body scans). Baseline documentation of ‘target’ and ‘non-target’ lesions When more than one measurable lesion is present at baseline all lesions up to a maximum of five lesions total (and a maximum of two lesions per organ) representative of all involved organs should be identified as target lesions and will be recorded and measured at baseline (this means in instances where patients have only one or two organ sites involved a maximum of two and four lesions respectively will be recorded). Target lesions should be selected on the basis of their size (lesions with the longest diameter), be representative of all involved organs, but in addition should be those that lend themselves to reproducible repeated measurements. It may be the case that, on occasion, the largest lesion does not lend itself to reproducible measurement in which circumstance the next largest lesion which can be measured reproducibly should be selected. Lymph nodes merit special mention since they are normal anatomical structures which may be visible by imaging even if not involved by tumour. Pathological nodes which are defined as measurable and may be identified as target lesions must meet the criterion of a short axis of ≥15mm. Only the short axis of these nodes will contribute to the baseline sum. The short axis of the node is the diameter normally used by radiologists to judge if a node is involved by solid tumour. Nodal size is normally reported as two dimensions in the plane in which the image is obtained (for CT scan this is almost always the axial plane; for MRI the plane of acquisition may be axial, saggital or coronal). The smaller of these measures is the short axis. For example, an abdominal node which is reported as being 20mm· 30mm has a short axis of 20mm and qualifies as a malignant, measurable node. In this example, 20mm should be recorded as the node measurement. All other pathological nodes (those with short axis ≥10mm but <15 mm) should be considered non-target lesions. Nodes that have a short axis <10mm are considered nonpathological and should not be recorded or followed. A sum of the diameters (longest for non-nodal lesions, short axis for nodal lesions) for all target lesions will be calculated and reported as the baseline sum diameters. If lymph nodes are to be included in the sum, then as noted above, only the short axis is added into the sum. The baseline sum diameters will be used as reference to further characterise any objective tumour regression in the measurable dimension of the disease. All other lesions (or sites of disease) including pathological lymph nodes should be identified as non-target lesions and should also be recorded at baseline. Measurements are not required and these lesions should be followed as ‘present’, ‘absent’, or in rare cases ‘unequivocal progression’. In addition, it is possible to record multiple non target lesions involving the same organ as a single item on the case record form (e.g. ‘multiple enlarged pelvic lymph nodes’ or ‘multiple liver metastases’). Response criteria Evaluation of target lesions Complete Response (CR): Disappearance of all target lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to <10 mm. Partial Response (PR): At least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters. At least a 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm. (Note: the appearance of one or more new lesions is also considered progression). Progressive Disease (PD): Stable Disease (SD): Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study. Special notes on the assessment of target lesions Lymph nodes: Lymph nodes identified as target lesions should always have the actual short axis measurement recorded (measured in the same anatomical plane as the baseline examination), even if the nodes regress to below 10mm on study. This means that when lymph nodes are included as target lesions, the ‘sum’ of lesions may not be zero even if complete response criteria are met, since a normal lymph node is defined as having a short axis of <10mm. Case report forms or other data collection methods may therefore be designed to have target nodal lesions recorded in a separate section where, in order to qualify for CR, each node must achieve a short axis <10mm. For PR, SD and PD, the actual short axis measurement of the nodes is to be included in the sum of target lesions. Table – Time point response: patients with target (+/– non-target) disease. Target lesions Non-target lesions New lesions CR CR CR CR Non-CR/non-PD Not evaluated No No No CR PR PR PR Non-PD or not all evaluated No PR SD Non-PD or not all evaluated No SD Not all evaluated Non-PD No NE PD Any Yes or No PD Any PD Yes or No PD Any Any Yes PD Overall response CR = complete response, PR = partial response, SD = stable disease, PD = progressive disease, and NE = inevaluable Appendix 11A: INNOVA-‘INITIAL MEASURMENT FORM’ RECIST CRITERIA Patient’s initials:………………………………………………..................... Age (years):………………………… Hospital Number of the patient:................................................................. INNOVA trial ID: ................................... Date of first measurement (dd/mm/yyyy)....................................... OVERVIEW TUMOUR SITES Method(**) Site involved Presence of ‘non-target’ lesions (0-no,1-yes,9-unk) (0-no,1-yes,9-unk) Comments Site of primary Lymph node Lung Liver Bone Brain Skin Other soft issue Other site Ascites Malignancy related Pleural effusion (0-no,1-yes,9-unk) INITIAL MEASUREMENTS (longest diameters) AND DESCRIPTION OF TARGET LESION Lesion Site Prior irrad. Date of measurements Measurements Method (*) (**) (dd/mm/yyyy) (mm) (##) Comments A B C D E F G H I J Baseline sum LD (Sum of the longest diameters): …………………………….. (*) Site: 1=Primary tumour/recurrence, 2=lymph node, 3=lung metastasis, 4=liver metastasis, 5=bone metastasis, 6=brain metastasis, 7=skin metastasis 8=other soft tissue metastasis, 9=other metastasis (**) Prior radiotherapy: 1=outside prior irrad. field/no radiotherapy, 2=within prior irrad field, 3=lesion previously irradiated (##) Method: (most reliable method used): 1=Clinical examination, 2=X-ray, 3=Ultrasound, 4=CT – scan, 5= Spiral CT – scan, 6=MRI, 7= Radionucleides, 8= Other, specify........................................................................................., .9= Not Done Date:....................................... Investigator’s signature ..................................................................................................... Appendix 11B: INNOVA-FOLLOW-UP MEASURMENT FORM RECIST CRITERIA Patient’s initials:……………………………………………….................... Age (years):………………………… Hospital Number of the patient:................................................................. INNOVA trial ID: ................................... Date of measurements(dd/mm/yyyy)....................................... OVERVIEW TUMOUR SITES Method(**) Response of non- New lesions target(##) (0-no,1-yes,9-unk) Comments Site of primary Lymph node Lung Liver Bone Brain Skin Other soft issue Other site Ascites Pleural effusion (##)- Response 1=complete response, 2=incomplete response/Stable disease, 3=progressive disease, 4=not evaluable MEASUREMENTS (longest diameters) AND DESCRIPTION OF TARGET LESION Lesion Site Date of measurements Measurements Method (*) (dd/mm/yyyy) (mm) (**) Comments A B C D E F G H I J Follow-up sum LD (Sum of the longest diameter) ………………………. (*) Site: 1=Primary tumour/recurrence, 2=lymph node, 3=lung metastasis, 4=liver metastasis, 5=bone metastasis, 6=Brain metastasis, 7=skin metastasis 8=other soft tissue metastasis, 9=other metastasis (**) Method: (most reliable method used): 1=clinical examination, 2=X-ray, 3=ultrasound, 4=CT – scan, 5= Spiral CT – scan, 6=NMR, 9= Not Done 7= Radionucleides, 8= Other, specify...................................................................................................... Overall response: (1=CR, 2=PR, 3=SD, 4=PD, 5= not assessable) .............................................................................................. Date overall response accessed (dd/mm/yyyy)....................................................................................................... Date:...................................... Investigator’s signature .................. ................................................................................... Appendix 12: INNOVA Pre-operative assessment form Part APatient’s full name: ………………………………………………….. Age: Hospital ID: ………………………… INNOVA ID: …………………………… Part B: Details of neoadjuvant treatment: Chemotherapy: Type of chemotherapy: Neoadjuvant □ Interval □ Date of last IV chemotherapy (dd/mm/yy): No. of cycles of chemotherapy: 3 □ 2 □ 1 □ 0□ If <3, specify reason Was there any Grade 3 or 4 adverse events during chemotherapy? Yes□ No□ Chemoradiation: Date of completion of chemoradiation (dd/mm/yy): Was there any deviation from protocol? Yes□ No□ If yes, specify reason………………………………………………………………………………………………………… ………………………………………………………………………………………………………………… ………………………………………………………………………………………. Part C: Clinical examination: Complete clinical response? Yes □ No□ (cCR only if no ulcer/nodularity/induration and sigmoidoscopy shows only whitening of mucosa or telangiectasia) If residual lesion present, distance from anal verge (cm): Serum CEA (ng/ml): Part D: Radiological assessment: Local (MRI) Maximum tumor length (mm) Maximum wall thickness (mm): Local invasion: Submucosa(T1) □ Muscularis propria (T2) □ Beyond muscularis propria (T3) □ Adjacent organs (T4) □ Maximum distance of residual tumor beyond muscularis propria (mm): No. of nodes ≥15mm in short axis: No. of nodes with diffusion restriction: Longest dimensions of metastatic node: Node 1(mm): Node 2 (mm): Distance of largest node/tumor from nearest mesorectal fascial margin (mm): MRF threatened? Yes □ No□ Distant Metastasis present? Yes □ No□ If yes, specify site Stage: ycT □ N □ M □ RECIST response: Complete □ Partial□ Stable □ Progression□ Completed by (name) ……………………………………………… Signature…………………………… Form completed on (dd/mm/yy) ……………………… Appendix 13: LOREC Histopathology Dissection and Reporting Protocol The pathology reporting in the LOREC programme is critical as the key outcome measures used to define success will be the rate of circumferential resection margin (CRM) involvement (defined as tumour cells 1mm or less from the inked CRM), intraoperative perforations (defined as a communication between the lumen and the external surface) and the planes of surgery of the mesorectum and the levator/anal sphincter. 1 Preparation of the specimen prior to dissection The intact specimen must be photographed whole prior to opening the bowel and further dissection. It is preferable for the specimen to be submitted fresh from the operation room to the pathology department as soon as possible after resection. Upon receipt in the pathology department, digital colour photographs should be taken of the anterior and posterior of the intact specimen. These photographs must include a ruler scale (for calibration), and the site of the tumour and the high vascular tie should be marked (e.g. with forceps or pre-printed labels). An example is shown below. If the tumour location cannot be ascertained by palpation, this should be indicated on the photograph with a label. Additional images of the lateral views, close ups of the anterior and posterior of the levator/anal sphincter (if appropriate), any perforation sites and any other unusual findings should also be taken. The plane of surgery should then be assessed by the local pathologist on either the fresh or fixed intact specimen (prior to opening) for both the mesorectum and the levator/anal sphincter area (as appropriate). This grading system is given below. The specimen can then be opened on the anterior peritonealised surface from the proximal resection margin down to a point approximately 2 to 5 cm above the level of the tumour. The mesorectum distal end should be kept intact, although the distal staple line can be opened to aid fixation. THE AREA OF THE TUMOUR MUST NEVER BE OPENED AS THIS DESTROYS THE ANTERIOR CRM. If fresh material is to be taken for local tissue bank use then it should be taken at this stage. A piece of foam/paper soaked in formalin can be inserted through the tumour if felt appropriate to aid fixation. The specimen should then be pinned out onto a cork board and placed in formalin fixative for approximately 48 hours. It is acceptable to inflate the specimen with formalin, leave to fix and then take the photographs, but this should always be before opening the specimen and prior to dissection. 2 Assessment of the plane of surgery The mesorectum and the area of the levator/sphincters should both be graded separately (where present). Thus for ARs specimen there will only be a grade for the mesorectum. For APEs there will be a grade for the mesorectum and a separate grade for the levator/sphincters below the mesorectum. The specimens should always be graded on the area of the 'worst' plane of excision. 2.1.1 Quality of resection of the mesorectum (all specimens) The quality of the mesorectal resection can be easily determined from the intact specimen and confirmed from the cross sectional slices. The three point grading system described below has been used in the MRC CR07 trial, MRC CLASICC trial and the Dutch TME/RT study, where poor planes were shown to predict a higher risk of local recurrence. Mesorectal fascial plane (good plane of surgery): The mesorectum should be smooth with no violation of the fascial covering. There should be a good bulk to the mesorectum both anteriorly and posteriorly, and the distal margin should appear adequate with no coning near the tumour. Any defect should only be very superficial and certainly not more than 5mm deep. Intramesorectal plane (moderate plane of surgery): There should be a moderate bulk to the mesorectum with irregularity of the mesorectal surface. Moderate coning of the specimen may be seen towards the distal margin. Importantly, the muscularis propria should not be visible, except at the area of insertion of the levator muscles. Moderate irregularity may be seen at the CRM. Muscularis propria plane (poor plane of surgery): There will be substantial areas of missing mesorectal tissue with deep cuts and tears down onto the muscularis propria. On cross sectional slicing the CRM will be very irregular and formed by the muscularis propria in places. 2.1.2 Quality of resection of the levator/sphincters (APE specimens only) The quality of surgery of the levator/sphincter area around the anal canal below the mesorectum should be separately assessed in APE specimens. Levator plane: The surgical plane lies external to the levator ani muscles which are removed en bloc with the mesorectum and anal canal. This creates a cylindrically shaped specimen with the levators forming an extra protective layer above the sphincters. There should be no significant defects into the sphincter muscles or levators. Sphincteric plane: Either there are no levator muscles attached to the specimen or only a very small cuff, and the CRM is formed by the surface of the sphincter muscles. There should be no deviations into the sphincters muscles themselves. The specimen shows coning at the level of the puborectalis muscle resulting in the classic surgical waist. Intrasphincteric/submucosal plane: The surgeon has inadvertently entered the sphincter muscles or even deeper into the submucosa. Perforations of the specimen at any point should also be classified into this group. 3 Macroscopic specimen dissection Once the whole specimen photographs have been taken and the plane of surgery graded, the fixed specimen is then ready to be dissected. The specimen should firstly be described in detail, with particular attention being paid to noting the presence of a perforation, and whether this is located at the tumour site or distant from the tumour. For perforations that involve the tumour site, it should be specifically stated whether the perforation is present in an area covered by peritoneum (TNM stage pT4) or an area of surgical margin (CRM positive), and additionally whether it is above or at the height of the sphincters All non-peritonealised surfaces (CRM) should be painted with ink (e.g. India ink) to facilitate the assessment of the CRM. Inking can be done before or after fixation according to local practice. It should be remembered that the CRM only applies to the surgically incised tissue planes and not the peritonealised surfaces. The CRM is larger posteriorly and extends up to a higher level than it does anteriorly (see image below). The specimen should then be cross sectioned into slices as thinly as possible (3-4mm is recommended) starting from the distal resection margin to at least the anterior peritoneal reflection or 2-5 cm above the tumour if higher. These slices should be laid out in good light starting with the most distal slice at the top left hand corner and the most proximal slice ending up as the last slice. Labels can be used to clarify the direction of the slices. The cut surface presented to the camera should be consistent in all of the slices. The slices should then be photographed either as a whole (with additional close ups of individual slices containing the tumour), or alternatively individual photographs of each slice can be taken. It is important that it is made clear on the photographs the order of the slices from the distal aspect of the specimen e.g. by using numeric labels. Again, the photographs must include a ruler scale for calibration. The slices should be carefully assessed after photography and the minimum distance of the macroscopic tumour to the inked CRM should be described, as should the maximum depth of invasion of the tumour beyond the outer muscle coat of the muscularis propria. This should be performed during the macroscopic assessment and confirmed histologically on the whole mount sections e.g. using the Vernier scale. If the position of the muscle layer is obscured by tumour or fibrosis, this should be estimated by comparison to subsequent slices. If the CRM appears to be free of tumour it should be noted whether there is normal tissue at the margin or whether it is fibrotic tissue suggestive of tumour regression. It is preferable to sample the primary tumour by embedding each tumour bearing slice in a large block and cutting a whole mount section. However, it is recognised that this is not possible in all laboratories so a minimum of 5 tumour blocks must be taken. All of the lymph nodes within the specimen should be identified and assessed, regardless of their site and size. A running mean of at least fifteen is to be expected in cases not undergoing preoperative therapy. The number of positive lymph nodes must be equal to or less than the number of lymph nodes sampled. The apical node (closest lymph node to the high vascular tie) should be identified and embedded separately to allow staging according to Dukes' classification. If lymph nodes lie close to or against the CRM then these should be included in the block where the relationship to the inked CRM will be maintained. 4 Microscopic reporting 4.1.1 Peritoneal involvement and extramural vascular invasion Involvement of the peritoneum should be carefully looked for and is defined as per Shepherd et al (see figure below). There should be a mean frequency of serosal involvement of 10% in rectal cancer specimens. Extramural vascular invasion is defined as involvement of a vascular structure which has smooth muscle in the wall. If tumour is present close to an arterial structure and the accompanying vein is not visible, then there should be a high level of suspicion for vascular invasion. There should be a mean frequency of extramural vascular invasion of 25% in rectal cancer specimens 4.1.2 CRM involvement Involvement of the CRM is defined as tumour cells at or within 1 mm of the inked CRM. If the tumour extends all the way to the CRM then the case is R1. If the tumour is greater than 0mm but less than 1 mm from the CRM then it is R1(<1mm) according to the revised TNM R1 guidelines. If the CRM is involved then the mode of involvement should be stated (e.g. primary spread, lymph node, tumour deposit, vascular, lymphatic, perineural etc). 4.1.3 pT staging of low rectal cancers The pT-staging of cancers above the sphincters is straightforward, however many low rectal cancers are partly located within the region of the sphincters. The anatomy of the levator/anal canal area is very complex and shows considerable variation between individuals. pT-staging of adenocarcinoma in the area of the sphincters is currently controversial. Both TNM 6 and 7 state that such tumours should be staged as anal cancers according to tumour size. However, in the absence of a robust evidence-based staging system, the only solution is to separately describe the anatomical extent of tumour spread both above the sphincters and at the height of the sphincters to allow subsequent analysis (see below). 4.1.4 Lymph node assessment and tumour deposits As stated in TNM 5, extramural tumour deposits measuring ≥ 3 mm in maximum size are counted as involved lymph nodes even if no residual lymph node structure can be identified. Smaller deposits are regarded as apparent discontinuous extensions of the main tumour. In the TNM staging system, pN1 corresponds to involvement of 1-3 nodes and pN2 to involvement of 4 or more nodes. 4.1.5 Preoperative chemoradiotherapy response scoring We recommend use of the Dworak method which is summarised below: 1. 2. 3. 4. No regression detectable Minimal regression: dominant tumour mass with obvious fibrosis and/or vasculopathy Moderate regression: dominantly fibrotic changes with few tumour cells or groups (easy to find) Good regression: very few (difficult to find microscopically) tumour cells in fibrotic tissue with or without mucin. 5. Total regression: no tumour cells, only fibrotic mass or mucin 4.1.6 Assessment of specimens where tumour cells are difficult to find Where tumour cells cannot be found on the first assessment of five blocks of tumour, the whole area of the tumour/scar should be embedded. If no tumour cells be seen following assessment of these extra blocks, then three deeper levels should be taken and examined from each tumour block. If after these assessments no tumour cells are identified then the tumour should be considered to have undergone a complete pathological response (TNM stage ypT0). Additional levels beyond the above should not be taken as it is important to standardise the degree of effort made to find residual tumour cells. 5 Definitions used in histopathological dissection and reporting 5.1.1 Position of the tumour The position of the tumour should be accurately noted on the histopathology report. This involves documentation of the quadrant of involvement from the cross sectional slices - i.e. anterior quadrant, posterior quadrant, lateral quadrant or combinations of these. To correlate the position of the tumour with the MRI report, the tumour should be described as a relationship to a clock-face from the distal resection margin with the peritoneal reflection being anterior (12 o'clock). ALL POSITIONS SHOULD BE REPORTED FROM THE PATIENTS PERSPECTIVE TO CORRELATE WITH THE MRI. 5.1.2 Relationship of the tumour to the anterior peritoneal reflection The crucial landmark for recording the height of rectal cancers is the anterior peritoneal reflection. This is identified on the exterior surface of the anterior aspect of the specimen. Rectal cancers are classified according to whether they are (see diagram below): 1. Entirely above the level of the peritoneal reflection anteriorly 2. Astride (or at) the level of the peritoneal reflection anteriorly 3. Entirely below the level of the peritoneal reflection anteriorly 5.1.3 Distance to the distal resection margin This is measured from the distal longitudinal cut-end of the specimen, not the CRM. It is only necessary to examine the longitudinal margins histologically if tumour extends macroscopically to within 30 mm of one of these. For tumours located further away, then it can be assumed that the cut ends are not involved. Exceptions to this recommendation are adenocarcinomas that are found on subsequent histology to have an exceptionally infiltrative growth pattern, show extensive vascular or lymphatic permeation, or are undifferentiated carcinomas. 5.1.4 Relationship to the dentate line This can only be measured for low rectal tumours in APE specimens. The dentate line should be defined as the level of the limit of the internal sphincter. As the distal rectum will have been cross sectionally sliced, this will have to be estimated on the basis of the slice thickness. 5.1.5 Tumour differentiation The differentiation of the tumour should be defined by the predominant area of tumour and not on the area of the worst grade. Other types of differentiation, i.e. mucinous adenocarcinomas, signet ring and undifferentiated should be documented. 6 Summary of data to be submitted 6.1.1 Histopathology report An anonymised version of the final signed-out histopathology report should be submitted. This should be identified by the unique histopathology report number, name of surgeon and date of operation. Macroscopic description including tumour site (above, at or below the peritoneal reflection), maximum tumour size, position of tumour (quadrants of involvement according to a clock face), distance from distal and proximal resection margins, presence and site of perforations, plane of surgical excision (mesorectal, intramesorectal or muscularis propria for the mesorectum and 6.1.2 extralevator, sphincteric or intrasphincteric/ submucosal for APE only), and distance from dentate line (for APE). Microscopic examination including type of tumour and grade of differentiation, local invasion (including depth of extramural invasion), margin involvement (including doughnuts, proximal and distal cut ends, distance to the CRM) and whether a complete (R0) resection. If the CRM is involved then report the maximal length of involved margin (measured histologically), mode of involvement (primary spread, lymph node, tumour deposit, vascular, lymphatic, perineural etc), type of tissue at CRM (tumour, fibrosis, normal). Any evidence of response to neoadjuvant therapy (if appropriate). Metastatic spread including lymph nodes (number retrieved and number involved, whether apical node involved), lymphatic or extramural vascular invasion, neural invasion, presence of extranodal deposits and histologically proven distant metastases. Co-existent conditions including ulcerative colitis or Crohn’s disease TNM (v.5) and Dukes’ stage. Digital photographs All of the digital specimen photographs (anterior, posterior, cross sectional slices and additional close ups) should be copied on to a CD/DVD and submitted. This is to allow blinded assessment of the quality of the plane of surgery. The site of the tumour and the site of the high vascular tie should be clearly marked (e.g. with forceps or pre-made labels) on all unopened specimen photographs. All photographs should always include a ruler/tape measure to enable calibration of the specimen. It may be advisable to ask the surgeon to take photographs of the fresh specimen following resection if this opportunity is likely to be missed in the pathology laboratory. Appendix 14: Timeline for activities for INNOVA Prior to entry Screening Physical examination Informed consent Randomization Blood tests Assessments Medical history Colonoscopy Histology MRI Abd/Pel CT chest ECG, Echo CXR Vital signs Height Weight DRE ICF X X X X X X X X X X X X CEA CBC RFT,LFT Pre-op investigations Prerandomization notepad Prerandomization radiology form RECIST initial form RECIST follow-up form Chemotherapy form ChemoRT form Pre-op assessment form PAC Surgery CRF Pathology form X X X Randomization Neoadjuvant chemo (all cycles) Pre During After neoadjuvant chemo ChemoRT Pre Post chemoR T Durin g Xa X Interval chemo (all cycles) Pre Durin g After interval chemo Xb Adjuvant chemo Pre Pre Afte r After adjuvant chemo Durin g X X X Xa Surgery X X Xb X X X X X Xc X X X X X X X X X X Xa Xb Xa X Xb X X a. Only in Arm A b. Only in Arm B c. Only WBC (during chemo); HB, Platelets once a week and WBC twice a week (during chemoRT) X X X X Appendix 15A: INNOVA Chemotherapy Form (Pre- Operative) Patient’s initials:………………………………………………...............INNOVA Trial No:………………………… Hospital Number of the patient:..............................................................Age(years): ................................... Height (m):..................................Oncologist: ................................... PART A Which chemotherapy regimen did the patient receive?Neoadjuvant □Interval □ PART B Did patient complete current 9 week cycle of chemotherapy?Yes □ □ No If No, reason?............................................................................................................................................. Date of cycle start: dd/mm/yy Cycle No.(for this regimen): Weight (kg) WBC Neutrophils Platelets Serum bilirubin (mg/dl) Estimated Creatinine (ml/min) Chemo given as: IP □ □ OP IP □ □ OP IP □ □ OP Actual dose given (In mg, not mg/m2): (If a protocol drug is omitted for any reason enter ‘0’) Daily capecitabine Mg/day Mg/day Mg/day Oxaliplatin mg mg mg Cycle Delayed* Dose(s) modified* *specify major reason(s) Capecitabine…………. Capecitabine…………. Capecitabine…………. Oxaliplatin………………. Oxaliplatin………………. Oxaliplatin………………. for dose delay/modification(s) using codes below Codes 0=No dose/delay modification4=Diarrhoea 8=Hepatobiliary dysfunctionC=Venous access problems Dose(s) reaction5=Hand modified* 1=Allergic foot syndrome9=Renal dysfunctionD=Capecitabine non-compliance 2=Neutropenia 6=Cardiac dysfunctionA=skin toxicity 3=Thrombocytopenia 7=Neurotoxicity E=Hospital administrative B=Stomatitis F=patient choice G=other, specify (including combination of reasons:................................................................................................................. ............................................................................................................................................................................................................ ...... PART C Toxicity Assessment Please complete after the end of pre-operative chemotherapy(neoadjuvant or interval) Date of assessment(dd/ mm/yy)………………………………….. Please record the worst CTC grade (CTC version 4.0) experienced during the pre-operative chemotherapy. Please record 9 if not known. □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ □ Nausea Pain Vomiting Stomatitis Lethargy Neutrophils Anorexia Alopecia Nail Changes Platelets Skin rash Peripheral neuropathy Haemoglobin Vein pain Diarrhoea WBC Hand-foot Other, specify........................................................................................................ Current WHO Performance Status □ 0=Able to carry out normal activity without restriction 1=Restricted in physical strenuous activity but ambulatory and able to carry out light work 2=Ambulatory and capable of self-care but unable to carry out any work. Up and about more than 50% of working hours 3=Capableonly of limited self-care,confined to bed or chair more than 50% of waking hours 4=Completely disabled, cannot carry out any self-care, totally confined to bed or chair Has the patient experienced any SAEs in the past 6 weeks? Yes □ No □ If Yes, has an SAE form been completed? If not, please complete immediately and inform the DSMB How many nights has the patient spent in hospital during preoperative Chemotherapy? (Please complete each field even if the entry 0) ICU.......................... chemotherapy....................... HDU............................ General/acute.........................In-patient How many dayshas the patient attended hospital during preoperative Chemotherapy? (Please complete each field even if the entry 0) Outpatient............................... Day Case................................. Appendix 16: INNOVA Chemoradiation form Part APatient’s initials: ………………………………………………….. Age (years): Hospital ID: ………………………… INNOVA ID:……………………………… Part B: Details of neoadjuvant treatment: Chemoradiation given as: IP □ OP □ Date of starting chemoradiation (dd/mm/yy):…………………………. Date of completing chemoradiation (dd/mm/yy):…………………………. Dose of radiation given (cGy):………………….. Daily Capecitabine dose:……………… mg/day Was there any deviation from protocol? Yes □ No □ (If yes, specify reason)…………………………………………………………………. Was the radiation pended for more than a week during course of treatment? Yes □ No□ (If yes, specify reason)…………………………………………………………………….. Part C: Toxicity assessment: Date of assessment (dd/mm/yy):………………………………. Please record the worst toxicity grade (Acute Radiation Morbidity Scoring Criteria) experienced during chemoradiation Skin □ □ WBC Mucositis □ □ Hemoglobin Lower GI □ □ Others (Specify) □ Genitourinary ……………………… Has the patient experienced any SAE during chemoradiation? Yes □ If yes, SAE form to be completed and DSMB to be informed No□ Completed by (name) ……………………………………………. Signature………………………… Form completed on (dd/mm/yy)…………………………. Appendix 17A: INNOVA Surgical Details: Intraoperative Form Patient ‘s initials:…………………………………………………… Age (years):………………………… Hospital No:................................................................. INNOVA No: ................................... Surgeon:……………………………………… Please answer ALL questions Part A Has there been any clinical evidence of ongoing toxicity Yes □ since completion of neoadjuvant treatment If ‘Yes’, No □ i) What was the toxicity? ......................................................................... Yes □ No □ Yes □ No □ ii)Was the patient admitted to Hospital? If ‘Yes’, Please complete SAE form iii) Was the surgery delayed? Part C Has the procedure resulted in a stoma? Y□ N□ End □ If ‘Yes’, was it: Loop □ Permanent □ Temporary □ Evidence of Metastasis: (Tick more than one if applicable) a) Liver: Yes □ No □ b) Omentum: Yes □ No □ c) Peritoneum: d) Other (specify): Yes □ No □ Yes □ No □ Y□ Serosal surface: Tumour evident on serosa? N□ N/A □ (if below peritoneal reflection) Macrosopic evidence of residual tumour following resection: Primary tumour? Nodes? Other? Intra-operative perforation If stapled anastamosis, level of anastamosis: Form completed by……………………………………. Date……………………………….. Uncertain□ Y□ N□ Uncertain □ Y□ N□ Uncertain □ Y□ N□ Yes □ At pelvic floor □ No □ Above pelvic floor □ Sign……………………………….. Appendix 17B: INNOVA Surgical Details: Hospital Discharge Form Patient ‘s initials:………………………………………………….. Age (years):………………………… Hospital No:................................................................ Date of surgery:………………………….. INNOVA No: ................................... N/A □ Date of hospital discharge* …………………………… * date fit for discharge, exclude time hospitalized for social reasons Complications-Please complete for all patients Part A Yes □ Did the patient experience any complications that required No □ prolonged hospitalization or were fatal or life-threatening? If ‘Yes’, please complete SAE form. Please √ one box for each question Part B Did the patient experience: Haemorrhage: Primary Yes □ No □ Yes □ No □ Yes □ No □ Wound Infection Yes □ No □ Intra-abdominal abscess Yes □ No □ Pulmonary complications: Yes □ No □ Deep vein thrombosis Yes □ No □ Urinary tract infection Yes □ No □ Death – complete SAE form Yes □ No □ Other complications If ‘Yes’, please specify Yes □ No □ Secondary Anastomotic leak If Yes, specify……………………………………. PART C Yes □ Was further abdominal surgery required? If ‘Yes’, please specify Form completed by……………………………………. PART D Date……………………………….. DINDO Grade: 0□ 1□ 2□ No □ Sign……………………………….. 3a □ Form completed by:…………………………………… Sign name............................................................... 3b □ 4a □ 4b □ 5□ Date completed:………………………… Appendix 18: INNOVA Pathologic Evaluation form PART A: Patient ‘s initials:………………………………………………................. INNOVA ID:............................................................... Age:………………………… Hospital No: ................................... Biopsy No:...........................................................Date of reporting: ................................... PART B: Gross Description - PART C:HISTOLOGY: Is there pathologic complete response?Yes□ No□ □ Surgery: APR □ □ □ □ ELAPE If No, type: Adenocarcinoma?Yes LAR ( Also tick box below if mucinous or signet ring adenocarcinoma ) Intersphinctric resection □ Mucinous Other (specify)............................................... - Plane of resection: Mesorectum : Mesorectal -Differentiation: poor signetring □ □ well/moderate □ - Maximum tumour thickness(mm)………………….. □ □ Intramesorectal Muscularispropria -Local invasion (pT stage): □ □ No T0 (pCR) □ For APE only: Levator plane(attached to mesorectum) Sphincter plane (on sphincter) T1(submucosa) □ □ T2(muscularispropria) □ Intrasphincteric / Submucosal /Perforation plane □ □ T3 (mesorectum) T4 (peritoneuminvolved/ □ □ tumour perforation/other organ) - Distal margin (cm) ............................................. - Proximal margin (cm).......................................... - Is there bowel perforation? Yes - Is there tumour perforation? Yes □ No □ N/A (if pCR) □ □ No -Maximum spread beyond muscularispropria (mm)…………. -If other organs involved, specify ................................ -Pathological nodal stage: □ No. of lymph nodesidentified .............................. No. of positive nodes ............................ Is apical nodal positive? Yes/No PN0 □ PN1(1-3 nodes) □ PN2(≥4) □ -Extramural intramural vascular invasion Yes □ □ No PART D: Resection margin PART E Is the CRM involved?Yes□No□ (tumour <= 1mm from involved circumferential margin) -If Yes, specify mode Direct tumour □ Node □ - Minimum distance from outer edge of tumour to CRM Tumour regression grade (Dworak) □ □ 0-No regression 1-Minimal regression (mm).............. 2-Moderate regression -Tissue at CRM (tick all that apply): Tumour □ Fibrosis □ □ □ 3-Good regression □ 4-Total regression □ Histologically confirmed liver metastasis? Yes Normal □ □ No Histologically confirmed peritoneal metastasis? - Is distal margin involved? Yes □ □ □ □ -Is proximal margin involved? Yes No : Yes □ □ No Pathological staging (TNM 7th edition): Type of resection: R0 □ R1□ (If no margin is involved) No ypT (If any margin involved) □ □ □ ypN ypM Additional Comments: ................................................................................................................................................................................................... ................................................................................................................................................................................................... Form completed by (name) :……………………………………………….................Date completed:………………………… Signature:............................................................... Appendix 19: INNOVA Serious Adverse Event Form Please report immediately any SERIOUS ADVERSE EVENTS by completing all of the details below. Please also complete the SAE form if the patient dies of any cause other than colorectal cancer. Patient ‘s name:……………………………………………… Hospital No................................................................. Age (years):………………………… INNOVA No: ................................... Responsible Onclogist……………………………………… SAE Description Is this an Initial or follow up report? Is this the final report? □ Y□ Initial □ N□ Follow up Reason for Reporting Death? Life threatening event? □ Y□ Y □Dateof death………………… N□ N In-patient hospitalization or prolongation of □ Persisitent disability or significant disability/incapacity? Y □ Other persistent reason for reporting Y□ existing hospitalization? Y □If yes,no of days?................ N□ N □ N If other , please specify: Date event started: Date event ceased: Details of adverse event(Pleaseattach copies of relevant details):........................................... ............................................................................................................................................ ............................................................................................................................................. Trial Treatment (This section must be completed by a clinician) Is SAE related to Chemoradiation □ Chemotherapy □ □ Surgery What was the date of surgery? If SAE SAE Description considered to be related to surgery, please assess causality (use codes given below):...................................................... Is this an Initial or follow up report? Initial Follow up If SAE considered to be related to chemotherapy: Is this the final report? Drug Date last dose Dose last given Reason for Reporting Administered Death? Life threatening event? Oxaliplatin In-patient hospitalization or prolongation of Capecitabine existing hospitalization? mg □ Y□ □ □ N No. of whole cycles given Y N □ Y□ Causality assessment (use Date of death………………… codes given below) □ N □ □ N □ If yes ,no of days?................ Persisitent disability or significant disability/incapacity? Y□ N □ If SAE considered to be related to chemoradiation: Causality assessment (use codes given below) Other persistent reason for reporting Y□ N □ mg Y If other , please specify: Last dose of capecitabine given: ........... mg Date last dose administered: ....................... Dose of radiation completed..................cGY Date last fraction given................................ Date event started: ……………………………… Date event ceased:…………………………. Details of adverse event (Please attach copies of relevant details):........................................... Causality assessment codes: ............................................................................................................................................ 1. Probably unrelated to treatment 2. Possibly related to treatment ............................................................................................................................................. 3. Probably related to treatment 4. Definitely related to treatment Please give reasons if you consider the event to be treatment related........................................ ...................................................................................................................................................................... .................................................................................................................................... Was the SAE unexpected? This section must be completed by clinician Unexpected □ Expected □ Please give reasons if you consider the event to be unexpected........................................................... ....................................................................................................................................................................... ................................................................................................................................................ Signature of Investigator:........................……… Name………………………………………..................... Telephone number:......................................... Date:………………………… Position:.............................