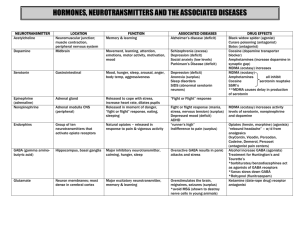
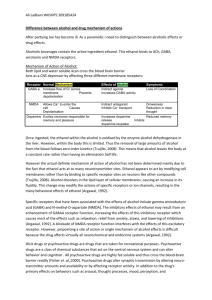
Risk assessment of MDMA - Utrecht University Repository
advertisement

Utrecht University
Risk assessment of MDMA
By: Laurens van Engelen, BSc. Supervisor Edwin
van den Worm, PhD.
“MDMA is the penicillin for the soul; you don’t give up penicillin when you see
what it can do.” (Alexander Shulgin, 1991)
1
Introduction
In this risk assessment, the question is not really whether 3,4-methyleendioxymethamfetamine
(MDMA) is toxic, since Paracelsus already stated: ‘all substances are poisonous, it is the dose that
distinguishes poison from remedy’. But can MDMA be used as a remedy as far as the toxicological
side of the drug is considered or is the therapeutic dose already a toxic dose? What is an acceptable
dose that can be considered acceptable risk if used for treatment of disease?
MDMA is used in tablets under the name Ecstasy by millions of people every weekend. In the
Netherlands, Ecstasy is the most used illicit drug after cannabis according to the federal drug monitor
(Van Laar 2012). From the experience of all these users it should be easy to acquire an acceptable
safe dose. However, retrospective experiments with Ecstasy users have shown to be very difficult.
First of all, there are the usual issues with retrospective research such as the so-called recall bias.
Secondly, most users do not know the exact concentration of MDMA in the tablets they ingested.
Thirdly, most Ecstasy users have experience with different drugs and may even use other drugs in
combination with Ecstasy.
In 2000, at the end of a Novartis symposium, the public was asked to write their opinion about the
proposed statement: “MDMA is a human neurotoxin”. The responses were mainly inconclusive with
the bigger part of responders being skeptical about the neurotoxic effects of MDMA under clinical
use (Turner & Parrott 2000). The responders also put forward that in cohort studies two very difficult
problems occur. First there is the recall bias: is what people call Ecstasy, MDMA or is it another drug
or ingredient that was used in the Ecstasy tablet? The second problem with this kind of research is
the problem of the control group that is used. It is hard to find a proper control group with as only
important variable the use of Ecstasy. Therefore this thesis does not make use of retrospective
research in order to determine the safety of MDMA.
2
Aim of this thesis
The therapeutic properties of MDMA were discovered by Alexander Shulgin in 1965, after which it
was used in more than a thousand therapy sessions in the United States of America. The high
potential for abuse of MDMA led to questions about illegalization of MDMA. Eventually, the
neurotoxic effects and the illicit use have led to the illegalization in 1988 and from then on MDMA
was classified as a schedule 1 drug by the DEA. This classification prohibits the therapeutic use of
MDMA and further testing in clinical trials. Therefore it was no longer possible to study or make use
of the potential medical properties of the drug.
During the last decade in Switzerland and other countries, clinical trials with MDMA have been
performed. This research is heavily criticized by some people who argue that even a single dose of
MDMA possesses neurotoxic properties (Parrott 2012).
Nevertheless, promising results have been found in a clinical trial on MDMA as a treatment for
individuals suffering from post-traumatic stress disorder (PTSD). In the light of those findings, a risk
assessment of MDMA is necessary (M. C. Mithoefer et al. 2013).
The dose of MDMA used in PTSD therapy is around 1-2 mg/kg body weight (BW) and will be
considered as the standard in the present study. The safety of this dose in the clinic will be assessed
via a scientific literature review. This review will treat MDMA as if it was a new drug to enter the
market. All use of ecstasy or MDMA as a party drug is beyond the scope of this risk assessment, and
therefore the emotions often related to the use of illicit drugs will not influence this review. Since the
‘war on drugs’ has tried to put all drugs in a negative ambience, this research pays extra attention to
the backgrounds and positions of the authors to make a well-weight risk estimation.
Disclaimer:
Any information given in this risk assessment cannot be used to justify the illicit use of MDMA.
3
Index
History
………………………………………………………………………………………………………………
Synthesis
………………………………………………………………………………………………………………
Use and addiction ………………………………………………………………………………………………………….
Pharmacology ………………………………………………………………………………………………………………
- Mechanism of action
Pharmacokinetic ……………………………………………………………………………………………………………
- Adsorption
- Distribution
- Metabolism
- Excretion
- Pharmacodynamics
Toxicity Risk assessment …………………………………………………………………………………………………
- Hyponatremia
- Hyperthermia
- Serotonin syndrome
- Hepatotoxicity
- Neurotoxicity
Discussion
Opinion
References
………………………………………………………………………………………………………………..
………………………………………………………………………………………………………………..
………………………………………………………………………………………………………………..
5
6
7
8
9
11
17
19
21
4
History
At the beginning of the 20th century, the search for CNS stimulants had started after the isolation of
ephedrine and epinephrine. Both substances can be obtained (naturally or synthetically) from
ephedra (Ephedra sinica), a plant that has been used for over 5000 years in Chinese medicine as Ma
Huang (Vearrier et al. 2012). The search for synthetic stimulants resulted in the development of
amphetamine-type substances, which were used for bronchodilation and nasal vasoconstriction
(Vearrier et al. 2012). After the first synthesis of methamphetamine from ephedrine in Japan, Smith,
Kline and French developed an inhaler for asthma and congestion called Benzedrine containing
methamphetamine in 1932, using ephedrine as a precursor (Vearrier et al. 2012).
MDMA was not synthesized as a stimulant, but as a precursor by Merck in 1912. In the development
of the new coagulation medicine named Hydrastinine, MDMA was an intermediate in the synthesis
and therefore it was patented by Merck in 1914 under the name “methylsafrylamin” (Capela et al.
2009). Merck tested MDMA twice; once in 1927 and once in 1959, but did not encounter any
psychoactive effects. In 1953, the University of Michigan secretly collaborated with the USA military
research program to test MDMA‘s toxicology. This brief historical overview may put some myths
about MDMA, such as that it was developed as an appetite suppressor and that MDMA was used in
World War One, to rest. However, methamphetamine was used in the Second World War by pilots
and soldiers. The myth that MDMA is an appetite suppressant comes from the substance MDA,
which is also a metabolite of MDMA (see below). MDA was tested in 1958 and patented by Smith,
Kline and French as an appetite suppressor. However, it never reached the market because of its
psychoactive properties (Parrott 2001).
Alexander Shulgin was the first to describe the psychoactive properties of MDMA. He synthesized the
drug and tested it on himself in 1965 (Agar & Reisinger 2011). For this reason Shulgin is often
regarded as the godfather of MDMA. In 1976, MDMA was first used in the clinic as an adjuvant to
psychiatric treatment by Leo Zeff (Capela et al. 2009). Shulgin and Zeff both successfully promoted
the use of MDMA in clinics for this property. In the eighties, MDMA was used under the name ADAM
in more than one thousand psychotherapy sessions in the USA. The dose used by the therapist varied
between 75 and 175 mg per session. While the use of MDMA by therapists in the USA was growing,
possesing, selling or distributing MDMA was illegalized in the UK in 1977 (Agar & Reisinger
2011)(Benzenhöfer & Passie 2010).
In 1980, a group in Texas started to sell MDMA in brown bottles under the name Sassyfras for
recreational use. The growing recreational use made the DEA start their procedure in 1984 to make
possessing and selling MDMA illegal. On July 27th of 1984, the DEA announced that they wanted to
schedule MDMA as a class 1 drug: a drug with no recognized medical use and which therefore cannot
be prescribed by a physician. A group of therapists and researchers requested a hearing. However,
the DEA was allowed to list MDMA as a schedule 1 drug without a hearing for one year. In this year
the DEA requested an international schedule 1 of MDMA by the WHO (World Health Organization)
which was completed at the 11th of February in 1986. At the hearing on May 22nd of 1986, judge
Young recommended to place MDMA in schedule 3, which would allow further research on the
compound. However, the DEA ignored the recommendation and on the 23rd of March 1988, MDMA
is permanently classified as a schedule 1 drug (Holland 2001).
All these trials drew the attention of newspapers and journalists and this rendered free publicity for
MDMA. The early name Empathy was changed to Ecstasy by the early drug dealers. The classification
of MDMA by the DEA ended almost all research and stopped the use of MDMA in psychotherapy.
However, the widespread attention enhanced the use of Ecstasy as a party drug. From 1987
onwards, the use of Ecstasy increased in the nightclubs of Ibiza where it was used on large dance
events. And since tourists widely shared their experiences with the drug throughout Europe, so did
the actual use Ecstacy (Holland 2001).
5
Synthesis
Both supply and demand of Ecstasy have grown big in Europe. Its production takes place
underground, for instance in abandoned labs in Eastern Europe. However, the Netherlands are
regarded as the major producer and distributor of the drug.(Holland 2001).
Figure 1 Synthesis route of MDMA starting with sassafras oil. PMK can be converted into MDMA (Gimeno et al. 2005).
The synthesis of MDMA starting with sassafras oil is relatively simple, but most syntheses of MDMA
start directly with substances that contain the methylenedioxyphenyl ring, such as safrole, piperonal,
isosafrole and piperonylmethylketone (PMK) as shown in figure 1 (Gimeno et al. 2005). PKM can be
converted into MDMA either via Leuckarts reaction or via reductive amination, the latter being the
predominant route (Gimeno et al. 2005).
Since law enforcement restricted the general access to safrole and piperol, new sources were found.
Piperol for instance, can be obtained from vanilla or pepperin, but the MDMA produced from these
precursors often has more impurities (Gallagher et al. 2012).
6
Use and addiction
As previously stated, MDMA was used as an enhancement in psychotherapy before its illegalization
in 1985 and was prescribed and used in therapy sessions over a thousand times (Parrott 2007).
Nowadays there is no good treatment for post-traumatic stress disorder (PTSD) while the
numbers of patients are quickly rising. Current treatment of PTSD consists of therapy together with
medication. However, drugs used in the treatment of PTSD are shown to have low efficiency. Recent
research shows that MDMA might be beneficial (Parrott 2007). The research into the use of MDMA
for psychotherapy has been delayed by legal issues and is only recently starting to re-develop.
(Parrott 2007). Research performed by Mithoefner (2010) has shown that therapy in combination
with MDMA (125mg and an extra dose of 62,5mg after 2 hours) is an effective treatment with a high
response in the treatment group of 83% versus 25% in the placebo group (M. C. Mithoefer et al.
2011). A follow-up for the randomized trial of Mithoefner showed that the results are persistent over
time and that MDMA would be a very effective treatment for PTSD (M. C. Mithoefer et al. 2013).
It is often thought that MDMA is addictive. However, unlike heroin, cocaine or crack use,
MDMA use does not lead to dependency as described in DMS-IV (Agar, 2011). Therefore, it is only
possible to describe the use of the drug. Most commonly, the patterns of use are mostly occasional,
not compulsive and in the order of once a week or once a month. Furthermore, users do not organize
their life around obtaining and using MDMA, contrary to users of heroin or crack (Agar & Reisinger,
2011). These observations are supported by statistics that describe that the use of MDMA depends
on the market: higher availability increases use and lower availability decreases use.
In animal experiments, addiction has been studied as well and investigators have shown that MDMA
increases dopamine levels in the Nucleus accumbens. However, the rats nucleus’ response to MDMA
is different from the response to cocaine or amphetamine since MDMA reduces the responding rate
to intracerebral self-stimulation. Interesting is the fact that D-amphetamine can substitute for
MDMA but the reverse is not true (Mohamed et al. 2011). This may support the statement that
MDMA is not as addictive as amphetamine.
The number of annual deaths in the United States due to MDMA use is 40 compared with 100,000
due to alcohol use. The fact that there is no harm reduction program such as (anonymous) pill
testing, flatters this statistic. While often 50% of the Ecstasy tablets do not contain MDMA or MDMAlike substances(Chang et al. 2000).
Therefore it may be concluded that the use of Ecstasy shows few indications of physical dependence
or drug craving. Moreover, a lot of users of MDMA stop after a while as they start to respond less to
the MDMA, this might influence the cost-benefit ratio in a bad way since the side effect increase and
the effect decrease. This is also seen in monkeys, that ceased responding for MDMA after an
extended period (approximately 2 years) of self-administration. However in the case of cocaine and
amphetamine this extinction is not observed. This is a further indication of the low risk of addiction
to MDMA (Parrott 2001; Mohamed et al. 2011).
7
Pharmacology
The chemical structure of MDMA has a lot of similarities with the structure of serotonin; therefore
MDMA can enter the cell via the serotonin transporter (5-HTT) (see fig 2). Serotonin regulates
behavior and attention in the nervous system and decreases body temperature via the
hypothalamus. Serotonin is formed
out of L-tryptophan by the enzyme
tryptophan
hydroxylase
(TPH)
located in the neurons, after which
serotonin is stored in granules in the
neurons.
Under
normal
circumstances
the
released
serotonin is transported back into
the synapse, via the 5-HTT, where it
is stored again in the granules via
the
vesicular
monoamine
transporter (VMAT). Free serotonin
can be metabolized by monoamine
oxidase (MAO-B) as can be seen in
figure 3 (M. Carvalho et al. 2012;
Figure 2 chemical structure of serotonin , MDMA, Tryptophan, and Dopamine
Capela et al. 2009).
have a lot of similarities
After MDMA has entered the cytoplasm of the cell, serotonin is released via two mechanisms (see fig
3). Firstly, serotonin molecules
exit the cell via the reversed 5HTT along the concentration
gradient
since
the
concentration
gradient
changed due to the entrance
of MDMA. Secondly, MDMA
uses VMAT to enter the
storage vesicles and depletes
the serotonin from the storage
vesicles by reversing the
transporter activity. Both
mechanism are based on the
structural similarity of MDMA
and serotonin, this results in
depletion of the free serotonin
from the neurons and second
for the storage vesicles inside
the neuron (Capela et al.
2009).
MDMA also partially inhibts
MAO-B, thereby preventing
Figure 3 The MDMA pharmacological mechanism of action at the neuronal
MAO-B which breaks down free
serotonergic terminal and synapse. 1) MDMA enters the neuron via the 5-HT
serotonin in the cell under
transporter (5-HTT). 2) Inside the neuron MDMA enters the storage vesicles via
normal circumstances. Leading
the vesicular monoamine transporter and releases the stored 5-HT. 3) MDMA
inhibits the enzyme tryptophan hydroxylase. 4) MDMA partially inhibits MAO-B. to an even higher serotonin
5) With the increased concentration of free 5-HT in the neuron the 5-HT is
concentration in the synapse
released out of the neuron via reversal of the 5-HTT. 6) MDMA binds to the 5(Capela et al. 2009).
HT2A responsible for the hallucinogenic effect of MDMA (Capela et al. 2009).
MDMA stops the production of
new serotonin irreversibly as it
8
inhibits TPH, which is the rate-limiting enzyme in 5-HT synthesis, irreversible. This inhibitions leads to
the breakdown of TPH which needs to be resynthesized after MDMA exposure, this explains a lower
serotonin level in the month following the use of MDMA(M. Carvalho et al. 2012).
MDMA has direct action on receptors as well, as it is an agonist to the postsynaptic 5-HT 2A receptor
leading to hallucinogenic effects. The main metabolite MDA has the same agonistic properties
(Capela et al. 2009).
Since MDMA leads to the releases of all serotonin and blocks the reuptake from the synapse and
stops the production of new serotonin, serotonin in the presynaptic cell can be depleted within 3 to
6 hours after MDMA administration. Because of the irreversibly inhibited TPH, the formation of new
serotonin in the following days is very low leading to a possible depression since the serotonin levels
are below baseline. However, during the serotonin boost the person experiences emotional
closeness and sensory delight, which may be very pleasant. Unfortunately, the serotonin boost also
produces an increased heart rate and blood pressure, tremors, excessive sweating, bruxism and
hypertension (Oesterheld et al. 2004).
The effects produced by MDMA are not all the result of serotonin since MDMA is not only a releaser
and reuptake inhibitor of presynaptic serotonin, but also of dopamine and noradrenalin. MDMA
inhibits the serotonin transport (5HTT), noradrenaline transporter (NAT), and the dopamine
transporter (DAT) with the potency of 5-HTT>NET>DAT (Capela et al. 2009). MDMA uses the same
mechanism that (meth)amphetamine uses, however the methylenedioxy ring has increased the
potency of MDMA for serotonin substantial, leading to the described serotonergic effect (De la Torre
et al. 2004;Oesterheld et al. 2004).
Pharmacokinetics
Absorption
MDMA is a weak basic drug and has a low molecular weight of 193.25 g/mol (M. Carvalho et al.
2012).
MDMA is usually ingested as a single dose of 100 to 200 mg. The maximal effect takes place after 6090 minutes (M. Carvalho et al. 2012). The precise bioavailability of MDMA in humans is unknown. A
low protein binding and a high volume of distribution ( 452± 137 L after 100 mg oral 6.4L/KG ) shows
the ability of MDMA to diffuse easy across cell membranes and lipid layers (Capela et al. 2009). The
Cmax is 1.5 – 3h up to a dose of 150 mg. The increase in Cmax and area-under-the-curve (AUC) for 24h
seem to be dose-dependent; however for a dose of 150 mg the increase is not proportional therefore
implying a non-linear pharmacokinetic profile. This could be explained by the metabolic saturation
and the ability of MDMA to inhibit its own metabolism via CYP2D6 (Capela et al. 2009).
Distribution
MDMA has a high bioavailability, a high volume of distribution and a low plasma-protein binding (M.
Carvalho et al. 2012).
Metabolism
MDMA is metabolized via two main pathways: either via O-demethylenation followed by catechol-Omethyltransferase (COMT)-catalyzed methylation and/or glucuronide/sulfate conjugation; or via Ndealkylation, deamination, and oxidation to the corresponding benzoic acid derivatives conjugated
with glycine (De la Torre et al. 2000). MDMA N-demethylation produces MDA. MDMA and MDA are
O-demethylenated to HHMA and HHA respectively. These HHMA and HHA can be O-methylated by
COMT creating HMMA and HMA (see fig 4).
9
Figure 4 Metabolism of MDMA and the CYP enzymes involved (De la Torre et al. 2000).
MDMA is mainly metabolized in the liver by different CYP450 enzymes with CYP2D6 being the most
important enzyme. These different enzymes give rise to inter-individual differences (Rietjens et al.
2012).
CYP2D6 is the main enzyme involved in the metabolism of MDMA and therefore individual
differences (polymorphisms) can play a role in MDMA-toxicity. The difference between ultra-rapid
metabolizers and poor metabolizers in blood concentration is expected, however since MDMA
inhibits CYP2D6, all patients act as a poor metabolizer after exposure (Green et al. 2012b). This
polymorphism varies between ethnics groups. Poor metabolizers have a bigger risk for severe
adverse effects such as described below, hyperthermia, hypertension, tachycardia, seizures,
serotonin syndrome, and rhabdomyolysis (muscle breakdown). However, poor metabolizers have a
lower risk to be exposed to the toxic metabolites. Inhibition of CYP2D6 by MDMA decreases the
formation of HHMA by 30-75%. MDMA is known to inhibit its own metabolism. This effect is seen at
recreational dose and the inhibition can be observed for several days with a recovery half-life of 2
days. A second exposure to MDMA in the days after use of MDMA will lead to a higher MDMA
concentration and therefore a higher risk of acute toxicity. Although re-exposure to MDMA soon
after the first exposure may be unlikely, the inhibiting effect on CYP2D6 can have severe
consequences for other medications that are metabolized via CYP2D6 . Since CYP2D6 is not the only
route of metabolism of MDMA, its inhibition may be compensated by other CYP enzymes such as
CYP2B6, CYP2C19, and CYP3A4 which are also capable of metabolizing MDMA. The different
polymorphisms in these CYP’s are another cause of broad inter-individual difference in the MDMA
concentration. An induction of CYP2B6, -1A2, and -2C19 could protect the liver since HHMA is more
hepatotoxic than MDMA, MDA, and HMA. However, the effect of MDMA would last longer giving the
10
problem of increased blood pressure
(Rietjens et al. 2012). However, since
these enzymes are less efficient
metabolizers, it takes longer to clear the
MDMA from the circulatory system, which
prolongs the period of increased blood
pressure.
Excretion
MDMA is eliminated for 80% after hepatic
metabolism while 20% is excreted
unchanged in urine. Half-life varies
between 6 and 12 hours (M. Carvalho et
al. 2012).
Pharmacodynamics
The best known effect of MDMA on the
body is the increase in 5-HT and to a lesser
extend dopamine in the brain. This is
caused by inhibition and reversal of the 5HT reuptake transporter (SERT). At the
same time, MDMA also influences the
VMAT, which is involved in the vesicular
storage of 5-HT (Rietjens et al. 2012).
The rate-limiting reaction in biosynthesis
of 5-HT is the hydroxylation of tryptophan,
which is catalyzed by tryptophan
hydroxylase. In rats, a single MDMA
exposure decreases TPH activity within
hours and can last up to several weeks.
This inhibition may be caused by the
metabolites of MDMA since MDMA was
not able to inhibit TPH in vitro (Capela et
al. 2009).
Figure 5 MDMA (▪) and HMMA (□) Plasma concentration (µg/L) after
oral intake of 50, 100, and 150mg MDMA one person per dose (De la
Torre et al. 2000).
Toxicity
The lack of toxicity of MDMA may be
illustrated by the few suicides commited
by ecstasy (Fernando et al. 2012). A blood
plasma level between 0,5 and 10 mg/L is
considered to be linked to fatality in most
cases (Kalant 2001). The plasma
concentration after oral intake of MDMA
are illustrated in figure 5, the increase of
the plasma concentration of MDMA is not
linear(De la Torre et al. 2000).
The LD50 for MDMA varies depending on
species. The LD 50 in mice is 80-115
mg/kgBW while the LD50 in rats was found
to by 18-42,5 mg/kg BW (Steele et al.
1994).
11
Hyperthermia
Body temperature is a complex balance between heat production and dissipation. The mechanism by
which MDMA influences the body temperature is complex since MDMA has effects on all major
monoamine neurotransmitters. Hyperthermia is considered to be one of the most life-threatening
acute effects observed after consumption of Ecstasy (M. Carvalho et al. 2012). All stimulant drugs
increase metabolic activity and therefore have the potential to induce hyperthermia (M. Carvalho et
al. 2012). The increase in body temperature by Ecstasy can lead to a body temperature of over 40
degrees Celsius. A high ambient temperature combined with intense dancing for many hours without
rest and without adequate hydration increases the body temperature even further. In some cases
the hyperthermia will lead to collapse or convulsions. Clinical examination will show dilated pupils,
sweating, a high heart rate (140-160), and low blood pressure with a core body temperature of 3942. These cases are in need of immediately medical attention. If the person does not stop his
activities in order to cool down, the hyperthermia may induce liver failure, kidney failure, cerebral
edema, all of which may lead to death (Kalant 2001).
Another dangerous complication with hyperthermia is rhabdomyolysis (muscle breakdown). This
phenomenon damages the kidneys and leads to high potassium levels, which can cause fatal heart
rhythms. Another commonly reported complication of hyperthermia is disseminated intravascular
coagulopathy (DIC): a blood clotting disorder that can cause the patient to bleed to death. When the
bleeding has started it is hard to control (Kalant 2001).
It has been show in animal experiments that the body temperature is associated with the damage
caused by MDMA making it even more unwise to use MDMA in hot ambient temperature. It has
been seen that people who take MDMA in psychotherapy session do not suffer from hyperthermia
(M. Carvalho et al. 2012).
Treatment of hyperthermia in the most severe cases starts with restoring the blood volume since
extracellular fluid volume tends to be depleted by the prolonged sweating. One liter intravenous
saline is given immediately, and if the pulse rate goes down and the blood pressure goes up, an extra
liter can be given. In milder cases the saline solutions will suffice, but in the more severe cases
individuals are actively cooled with for instance an ice bath. Dantrolene, a calcium antagonist, can be
administered to reduce the heat production from muscle contraction (Holland 2001). However, there
are no effective single pharmaceutical agents that reverse the hyperthermic effect (M. Carvalho et al.
2012).
Hyponatremia
A possible side effect of Ecstasy use is acute hyponatremia: a low plasma sodium level due to dilution
of the blood with water. This occurs after too much water consumption without enough mineral
intake. Hyponatremia is observed in users who are aware of the risk of dehydration and
hyperthermia. These users drink to much water to prevent dehydration (Capela et al. 2009).
MDMA increases the production of the antidiuretic hormone ADH in the plasma. ADH inhibits
urination and promotes increased water reabsorption in the kidneys. This increased reabsorption
together with an increased water intake may lead to a dangerous low sodium level in the serum. The
first signs are mute states, headache, and vomiting. Hereafter, nausea cramps, weakness, fatigue,
confusion and even seizures may follow. The worst possible symptom is cerebral edema: swelling of
the brain with water, eventually leading to death. The occurrence of hyponatremia is far lower than
that of hyperthermia. Treatment starts with stopping all fluid intake. In more severe cases hypertonic
saline can be given intravenously to restore the mineral balance (Kalant 2001).
12
Serotonin syndrome
The classic symptoms of serotonin syndrome are mental status changes, autonomic hyperactivity and
neuromuscular abnormalities as is shown in table 1, but not all the symptoms are observed in all
patients. The word ‘syndrome’ is misleading as it suggests an idiosyncratic reaction, whilst it is known
that serotonin syndrome is the result of concentration-dependent action of the serotonin at the 5HT2A receptor. Therefore, some articles prefer to use the term serotonin toxicity (Sun-edelstein et al.
2008).
Severe life-threatening cases are characterized by rigidity, decreased arterial carbon dioxide tension
(PaCO2) and fevers from 38.5 °C which can increase to over 41 °C. Patients may develop a shock if
the rigidity affects the truncal musculature.
The prognosis after serotonin syndrome is good as long as there are no secondary complications and
no long term neurological damage is observed (Sun-edelstein et al. 2008).
The serotonin syndrome is caused by drug-induced excesses of intra-synaptic serotonin, acting at
serotonin2-receptors. Serotonergic drugs such as Ecstasy as well as selective serotonin reuptake
inhibitors (SSRIs), tricyclic antidepressants (TCAs), and mono-oxidase inhibitors (MAOI) are known to
be able to induce serotonin syndrome (Birmes et al. 2003). A combination of these drugs increases
the risk on developing the serotonin syndrome. The more severe serotonin syndrome cases (i.e.
resulting in death) are not caused solely by SSRI, but most often by a combination of MAOI and SSRI
or a combination of MAOI and amphetamines (Sun-edelstein et al. 2008).
The use of MDMA in combination with these antidepressants is a cause for the development of
serotonin syndrome since there is a cumulative effect leading to levels of serotonin higher then when
the drugs are used in the form of a single dose. However it is important to note that SSRIs block the
uptake of MDMA by the serotonergic neuron and therefore lower the effect of MDMA. This has been
shown in a clinical trial, wherein patients were first administered Citalopram, a SSRI, and
subsequently MDMA. The effect of MDMA was lower than without the pre-exposure to citalopram.
Therefore, it can be expected that the use of MDMA by patients who take chronic SSRIs do not have
an increased change on developing Serotonin syndrome. However, the administration of SSRIs after
the exposure to MDMA to a SSRI naive patient may give an increased risk (Liechti & F X Vollenweider
2000).
Neuromuscular hyperactivity
Akathisia (inability to sit still)
Tremor (unintentional rhythmical alternating movement)
Clonus (continuous tremor)
Myoclonus (sudden twitching of muscles)
Hyperreflexia (overresponsive reflexes)
Rigidity (resistance to passive movement)
Nystagmus (involuntary eye movement)
Autonomic hyperactivity
Diaphoresis (excessive sweating)
Fever
Tachycardia (heart rate over 100 beats per minute)
Tachypnea ( rapid breathing)
Altered mental status
Agitation (unintentional and purposeless motions)
Excitement
Confusion (being unclear in one’s mind about something)
Table 1 Clinical features associated with Serotonin Syndrome (Sun-edelstein et al. 2008).
Treatment of serotonin syndrome depends on the severity of the symptoms and the nature of the
drug ingested. The first treatment of patient with serotonin syndrome is sedation and active cooling.
13
Further treatment consists of removing the drugs, administration of 5-HT2a antagonists and gaining
control of agitation, autonomic instability, and hyperthermia. Paralysis can be induced by
administrating non-depolarizing agents in severe cases. Neuromuscular paralysis is the accepted
treatment for hyperthermia resulting from excess muscle activity (Holland 2001)(Sun-edelstein et al.
2008).
This syndrome is consistent with a mortality rate between 10 and 15%. This percentage is
biased since the less severe cases are not reported. The occurrence of serotonin syndrome may
explain the cases observed with hyperthermia without minimal physical exercise. Therefore it can be
difficult to distinguish patients with serotonin syndrome and those with hyperthermia (Holland
2001).
Since MDMA releases serotonin and inhibits its reuptake, it is expected that the increase of serotonin
overstimulates the 5-HT2A receptor which lead to serotonin syndrome. It is expected that the
occurrence of mild serotonin syndrome is probably high but under-diagnosed in MDMA users. Even
though severe serotonin syndrome will not develop from normal MDMA use, the use of other
serotonergic medication may result in severe serotonin syndrome which may end in death (Holland
2001).
Hepatotoxicity
A serious adverse reaction to Ecstasy use is hepatitis: inflammation of the liver. There have been
several cases reported but these cases were inconclusive to define MDMA as the inciting agent. In
studies with rat an dog no hepatotoxicty has been observed. Therefore it is thought that liver
damage is an idiosyncratic drug reaction. Therefore it is not dose dependent and highly
unpredictable(Holland 2001; M. Carvalho et al. 2012).
Neurotoxicity
The most discussed side effect of MDMA is its neurotoxicity towards serotonergic neurons. Ever since
the first report by Schmidt et al. in 1986 which suggested that MDMA is a neurotoxin, this discussion
has further evolved (Capela et al. 2009). Schmidt et al suggested that MDMA has neurotoxic effects
on serotonergic neurons since the serotonin level is lower seven days after exposure to MDMA, this
is a result from the inhibition of THP as described before. A high number of animal experiments do
support the expected MDMA’s neurotoxicity in animals. However, the translation of results obtained
in animal studies to a human population remains very difficult. The animal experiments used high
doses of MDMA for periods up to several days. As previously described in the section about use,
MDMA in humans will only be used a few times in psychotherapy.
Researchers are skeptical about these animal tests and the subsequent translation of the obtained
results to the human situation. If animal research finds a dose-depended curve for neurotoxicity, this
certainly cannot be directly applied to humans, since the dose used and the dosage regime are far
out of the reach of the recreational use as will be discussed further below (Turner & Parrott 2000).
To go back to the criminalization of MDMA in 1985, Ricaurte has always been against the use of
MDMA (McCann et al. 2000). It is interesting to note that Ricaurte had to retract one of his most
startling experiments in whichin he proved severe dopaminergic neurotoxicity in non-human
primates after a recreational dose regime of MDMA. He had to retract his article because the MDMA
he used in this study was accidently switched with methamphetamine (George A Ricaurte et al.
2002). Although it is not clear what exactly has gone wrong, but Ricaurte claims that the
manufacturer has switched the MDMA and the methamphetamine. However, this incident made his
peers more cautious about the credibility of the rest of his research
In Ricaurtes article on MDMA induced neurotoxicity (2000) it is stated/claimed that a dose of 5 mg
already leads to neurotoxicity in non-human primates (G A Ricaurte et al. 2000). The exact dose
turned out to be 5 mg/kg, as will be further discussed in the next paragraph. Such a sloppy mistake is
remarkable at least, considering the impact and importance of his statement. Furthermore, the dose
that he mentioned originates from one of Ricaurtes earlier publications (1988). The concentration of
serotonin had been measured by HPLC, two weeks after exposure to MDMA. As mentioned
14
previously, the irreversible breakdown of TPH by MDMA will take more than a month to restore,
which is a possible explanation for the lower serotonin levels aside from neurotoxicity. Furthermore,
the experiment was based on a sample size of n=3, which is very low for such a study and therefore
should always evoke caution.
In Ricaurtes second argument he extrapolated the so-called neurotoxic dose of 5mg/kg BW to
humans and claimed it to be 1.28 mg/kg BW. In the next paragraph, the article of Green will be
discussed in relation to the extrapolation of Ricaurte. However, going back to the research of 1988:
this is only suggesting that a dose of 1.28mg/kg BW in humans can lead to a decrease of serotonin in
the brain as long as two weeks after exposure. As previously stated, this does not have to point
towards neurotoxicity but may simply be explained by TPH activity.
The author declared no conflict of interest and acknowledged that the research is paid from 3 grants
which were all three received from U.S. Department of Health and Human Services Public Health
Service. All together this particular review is very weak and does not prove the toxicity for humans.
One of the most up-to-date discussions is the article from Green (2012) and the commentary written
by Parrot who both do declare to have no conflict of interest. Both are experts on MDMA and its
neurotoxicity. The review of Green is named “Lost in translation: preclinical studies on MDMA
provide information on mechanisms of action, but do not allow accurate prediction of adverse events
in humans”(Green et al. 2012b). Parrot responded with a commentary “MDMA and 5-HT
neurotoxicity: the empirical evidence for its adverse effects in humans – no need for
translation”(Parrott 2012).
Green is in favor of using MDMA as medicine, while Parrot thinks any consumption of MDMA should
be illegal. Green states that MDMA has been shown to be neurotoxic in laboratory animals, but that
extracting a safe dose from the animal experiments has little value since the long term neurotoxicity
is mainly a result of metabolites. Since the MDMA metabolism in humans is completely different
from that of rats or primates, data derived from animal experiments cannot be used to estimate a
safe dose. Green et al. use pharmacokinetic-pharmacodynamic integration to translate preclinical
data into data that can be used in clinical experiments. Green et al.. started with the exposure, the
dose of 5 mg/kg BW in a rat will be around 350 mg for a person of 70 kg based on a one on one
translation of mg/kg BW. Ricaurte used a allosteric translation which led to a dose of 70 mg of
MDMA for an average person, however Green states that this measurement cannot be used when
the mother-compound is converted into active metabolites as is the case with MDMA. Human
experiments put forward that the dose-plasma concentration is not linear since there is a fourfold
increase in plasma concentration after only a double dose (1mg/kg to 2 mg/kg) which is due to the
auto inhibition of CYP2D6 by MDMA. MDMA is more rapidly absorbed and metabolized in rats than
in humans. Adding to this is that the percentage of MDA differs between rat (24%) and human (10%).
This difference together with the faster metabolism in rat gives rise to a higher MDA exposure in rat.
MDA is a known neurotoxic and may explain the neurotoxicity found in the rat. Importantly, the
effect in humans fast metabolizers is lowered since MDMA inhibits CYP2D6 and therefor all humans
somehow act like a poor metabolizer. Another point put forward by Green is that the route of
administration is practically ignored by researchers, since the MDMA was injected intravenously in
almost all experiments. This results in a threefold increase in AUC and in a fourfold increase in Cmax
and Tmax, compared with oral administration. Another point that was put forward by Green is that
there is no information about drug-protein binding available for rat or humans while this can have
important effects on exposure difference in humans and rat (Green et al. 2012b).
Green concluded that there is a substantial temporal mismatch for rat to human extrapolation for
long term neurotoxicity. Most important is that direct injection of MDMA into the brain of rats does
not lead to neurotoxicity and therefor it looks clear that the neurotoxic effect is probably a result of a
metabolite such as MDA. Green concludes that the doses currently used to investigate therapeutic
effects of MDMA, are unlikely to produce any severe acute or long lasting neurotoxic effects. Parrot
wrote a commentary on Green’s article putting forward that there is a lot of research proving that
Ecstasy is neurotoxic, which means no translation of animal to human is necessary. Green responded
15
by highlighting that his research is about the translation of animal research of MDMA, not Ecstacy, to
humans. He emphasized that Ecstasy cannot be considered to be the same as (pure) MDMA and that
the therapeutic use of MDMA cannot be compared with the illicit use of Ecstasy (Green et al. 2012a).
Another proposed mechanism behind neurotoxicity is the formation of free radicals which are toxic
to neurons. There is direct evidence that MDMA increases the free radical formation in the brain.
Colado et al. proved this in 1997 by measuring the conversion of salicylate to 2,3-dihydroxibenzoic
acid in the hippocampus via a micro dialysis probe. The conversion of salicylate is a reaction that is
driven by free radicals (Green et al. 2003).
In 1999, Aguirre et al. showed that administration of a high dose of antioxidant (α-lipoic acid) fully
protected against the damage to 5-HT nerve endings, which supports the theory that free radical
formation after MDMA exposure may be responsible for the neurotoxicity. Colado et al.1999
observed that when hyperthermic responses were prevented, the free radical formation was
inhibited(Green et al.. 2003).
In a way the formation of free radicals is a result of the disruption of the serotonin functions.
Carvalho et al. showed that the increase of serotonin in the terminal endings results in oxidative
stress via two mechanisms: auto-oxidation and the metabolism of monoamine. Both of these
processes can produce ROS. The body temperature influences the formation of ROS and therefore
the neurotoxicity. Malberg and Seiden (1998) proved that rats treated with 20-40mg/kg BW MDMA
at low ambient temperature (20-24⁰C) did not develop neurotoxicity while this still is the case with
rats living in higher ambient temperature (26-30⁰C)(Parrott 2001). Carvalho et al. (2012) suggested
that the substances which have proven to be neuroprotective simply inhibit the hyperthermic
response of MDMA.
Liechti (2001) tested MDMA in 58 humans, and in this study the body temperatures rose significantly
from 36.6 to 37 °C. However this is only a slight increase and certainly no sign of hyperthermia
(Liechti et al. 2001). So the body temperature in humans is no concern for the use of MDMA in
clinics.
16
Discussion
As previously described, MDMA is able to induce different side-effects. Hyperthermia is observed in
the illicit use of MDMA, mainly when users dance vigorously for hours in a hot environment. This is
the opposite from the use of MDMA in therapy where the patient would be sitting in a session. This
could be observed in clinical trials where there was only a slight rise in temperature seen in the
patients. Therefore it can be expected that hyperthermia is a side-effect primarily associated with
the illicit use of MDMA and not the therapeutic use. The same holds true for the other described
symptom of hyponatremia after an overdose of water. This side-effect is a result of the lack of
information during illicit use of MDMA and is also unlikely to occur during the therapeutic use.
The serotonin syndrome is hard to diagnose as this disease is not so severe. It is to be expected that
with the release of the serotonin in MDMA users, a light serotonin syndrome may occur. However, at
this level the serotonin syndrome is not harmful and people will recover within 24 hours. In
therapeutic use, care should be taken with the use of some antidepressants, as they might induce
serotonin syndrome when taken together with MDMA. This does not hold true for SSRIs. The chronic
use of SSRIs will probably extend the duration of MDMA, and therefore it would be unwise to
consume a second dose. The inhibition of CYP2D6 by MDMA is important information for the
pharmacist if a person wants to join a therapy session with MDMA. Inhibition of CYP2D6 will affect
the metabolism of other medication used.
Some of the metabolites of MDMA might have hepatotoxic effects, however no severe reactions
were detected in the clinical trials. A severe reaction on MDMA is the inflammation of the liver. This
is probably an idiosyncratic drug reaction and therefore more research will be necessary.
Idiosyncratic drug reactions are very rare but often very severe. Since its rareness, MDMA can still be
used as medicine however special attention should be paid to liver problems soon after exposure to
MDMA.
MDMA is a neurotoxic in animals; after exposure of a high dose for multiple days degeneration of
neurons can be seen. Whether these neurotoxic effects can be translated to humans remains difficult
to say. If MDMA is very neurotoxic to humans millions of people every week will be at risk when
(ab)using MDMA. However, the therapeutic use of MDMA takes place under ideal and controlled
conditions. The patient is at rest and does not overheat due to physical exercise. The dose is
controlled and the patient is watched over by the therapist. The research proving that ecstasy is
neurotoxic does not hold much validity for the therapeutic use of MDMA. First of all, the use of
ecstasy does not automatically mean that MDMA is consumed. People referring to ecstasy think they
use MDMA. However, depending on the country, a lot of ecstasy tablets do not or hardly contain
MDMA. To make things worse, if there is MDMA in the tablets the concentration is completely
unknown to the consumer. Adding to these problems is that the users of ecstasy often use several
legal or illegal substances such as caffeine, alcohol, cocaine and amphetamine which have significant
interactions with MDMA and are definitely affecting the possible neurotoxicity. This interferes with
retrospective research, especially as most of these other substances may not be known by the
researchers. Therefore retrospective research regarding ecstasy cannot be used in this risk
assessment.
Animal data are not troubled by possibly clouded information as is the case with retrospective
research, but still care needs to be taken. Experiments or researchers might be biased or make
mistakes as is seen with Ricaurte’s research wherein MDMA was accidently switched with
methamphetamine. And in his review about neurotoxicity of MDMA he referred to experiments with
N=3 to prove the toxicity of MDMA. However, as discussed by Green, the use of animal experiments
remains questionable. There is a big problem because of the difference on pharmacokinetics and
pharmacodynamics between species. Since MDMA is converted to the very active metabolite MDA
which is neurotoxic, the effect seen in animals might be due to exposure to the metabolite instead of
MDMA. This seems to hold true for rats which metabolize MDMA very fast compared to humans and
also produce more than 2 times more MDA than humans. Therefore the exposure of rats to MDA is
17
much higher than the exposure of humans to MDA. This gives rise to a complete different
toxicological profile, since MDA is a known neurotoxic compound.
The formation of free radicals might be the reason for the neurotoxic effect seen in animals.
However the free radical formation seems to be increasing with the temperature and while this is
concerning for the illicit use of MDMA it does not seem to influence the therapeutic use of MDMD as
will be discussed next.
The most valuable research in order to clear up the toxicity of MDMA is placebo controlled double
blind clinical trials. But since MDMA has been scheduled as a class one drugs this research has been
difficult for a long time. In the last decade some research has been performed,most notably in
Switzerland where the group of Liechti and Vollenweider has tested MDMA placebo controlled and
double blind on 74 volunteers. These results were very promising since the dose used in the
experiments is comparable with the dose used in therapy. These experiments showed only a slight
increase in temperature and therefore patients in therapy seem to be pretty much protected from
free radicals formation. As for the other effects seen in the patients, all the individuals seemed to be
very well and no severe complications have occurred. In recent experiments in 2010 MDMA was
tested for the use in treating PTSD and with some very promising results(M. C. Mithoefer et al. 2013;
M. C. Mithoefer et al. 2011). In the test group the number of patients that recovered was three
times higher compared to the placebo control group. Since these human trials seem so promising
and very severe toxic effects of MDMA would have been observed in the abuse of MDMA in the last
30 years, my personal opinion is that the therapeutic use of MDMA should be allowed. The emotions
involved with illicit drugs and the war on drugs should not hold back people from an effective
treatment. It seems that most dangers with ecstasy are not involved in the therapeutic use of
MDMA.
Conclusion
MDMA should be tested further for its properties as a promising drug in the treatment of PTSD. The
toxic profile of MDMA should not be a hold back especially not with the low frequency of use of
MDMA in PTSD. However, chronic exposure to MDMA might have different toxicological effects.
18
Opinion
Before this thesis, I believed as a pharmacist that the changes of a simple molecule administered
orally once, especially since it has been found ‘safe’ by a lot of users, will do no harm. I still do believe
this. Long-term changes will require more than one tablet since millions of people have used MDMA.
A lot of researchers will possibly disagree as they have found data suggesting a change after one
single administration. If a single tablet could change our brain, this would still be measurable after six
months. I believe the human body has enough repair and safety mechanisms in place to prevent this.
The only exception to this is idiosyncratic drug reactions which is a complete different category.
It is often forgotten that Alexander Shulgin, who discovered the psychoactivity of the drugs,
started giving it to therapists who in turn used it in over a thousand therapy studies. If MDMA had
been registered at that time, the whole discussion would have been completely different. A good
modern day example is Ritalin which was only abused after its registration for the treatment of
ADHD. There would not be discussions whether it has to be made illegal again since it already would
have proven to work. MDMA has the unfortunate problem that it was not registered and even
scheduled as a class 1 drug, which blocked all the research necessary to get it registered. The judge
involved in the illegalization already suggested to put MDMA in class 3 which would allow further
research. However, the DEA and their war against drugs were insensitive to this argument. In my
opinion the DEA considers the loss of therapeutic properties of MDMA as collateral damage in their
war on drugs. If the abuse of MDMA would entail very severe consequence for the user and
illegalization could make the drugs disappear, then it could be justified to claim that it would help
more people to make MDMA illegal. But MDMA is widely available and (ab)used and the
consequences appear less severe than thought on beforehand. However, the illegalization keeps
MDMA out of reach of those who could benefit from it. The research supporting the toxicity of
MDMA is in my opinion very weak since most only holds true for the illicit use of ecstasy and not for
the therapeutic use of MDMA. A good example of this is the research of Ricaurte putting forward the
toxicity of MDMA with animal experiments. But the translation of these experiments into a human
situation is very difficult. The same researcher has proven that the use of ecstasy is toxic. However,
this is not relevant to MDMA for multiple reasons. First of all, the composition of many ecstasy
tablets is not the same as MDMA and therefore the research cannot be translated to MDMA. Since
ecstasy is purchased illegally there is hardly any quality control or control on dose or substance
present. This is often shown with confiscated ecstasy tablets. The majority of these pills did not even
contain MDMA as the active substance. Adding to this problem is that the research performed on
active ecstasy users is troubled by all the other substances, whether legal or illegal, that are
consumed in combination with ecstasy. Second of all, as put forward in this risk assessment, the illicit
use of MDMA is troubled by flawed information to the public leading to unwise and possible unsafe
MDMA intake. People either drink too little water and get overheated, or are afraid to drink too little
and therefore get hyponatremia. The rave parties where ecstasy is consumed are often too hot and
relaxing areas are missing adding to the problems when people dance for hours continuously. My
opinion about the illicit use of ecstasy and- or MDMA has changed after this review. I think that there
is enough evidence that the illicit use of ecstasy combined with the unwise behavior of the people is
toxic. However, I believe that the illegalization of MDMA in fact has worsened the toxicity. I do not
believe that the use of ecstasy will be more harmful than the use of for example alcohol. A positive
characteristic is the lack of addiction of MDMA usage. If, instead of the war on drugs against ecstasy,
an efficient harm control program would be in place, as is the case with alcohol, the toxic effects of
ecstasy could even be further reduced. The ambient temperature at rave parties could be controlled
and relax areas could be placed. The right information would lower the amount of cases of
hyperthermia and hyponatremia. However, the arguments for the legalization of MDMA as a party
drug are beyond the scope of this research. The therapeutic use of MDMA lacks the abovementioned problems of illicit MDMA use. Since the dose in therapy sessions is controlled and often
as few as three therapy sessions are enough to help the patients, the total exposure to MDMA in
patients is far lower than in people who illicitly use MDMA. Furthermore, because of the relaxed and
controlled environment in which the drug is consumed, the patient is more likely to remain calm.
19
Also, medical attention is directly available. Therefore, I conclude that the use of MDMA in therapy is
completely legitimate from a toxicological perspective.
20
References
Agar, M. & Reisinger, H.S., 2011. Ecstasy : Commodity or Disease? Journal of Psychoactive Drugs,
36(2), pp.253–264.
Benzenhöfer, U. & Passie, T., 2010. Rediscovering MDMA (ecstasy): the role of the American chemist
Alexander T. Shulgin. Addiction (Abingdon, England), 105(8), pp.1355–61.
Birmes, P. et al., 2003. Serotonin syndrome: a brief review. CMAJ : Canadian Medical Association
journal = journal de l’Association medicale canadienne, 168(11), pp.1439–42.
Capela, J.P. et al., 2009. Molecular and cellular mechanisms of ecstasy-induced neurotoxicity: an
overview. Molecular neurobiology, 39(3), pp.210–71.
Carvalho, M. et al., 2012. Toxicity of amphetamines: an update. Archives of toxicology, 86(8),
pp.1167–231.
Chang, L. et al., 2000. Effect of ecstasy [3,4-methylenedioxymethamphetamine (MDMA)] on cerebral
blood flow: a co-registered SPECT and MRI study. Psychiatry research, 98(1), pp.15–28.
Fernando, T. et al., 2012. Ecstasy and suicide. Journal of forensic sciences, 57(4), pp.1137–9.
Gallagher, R., Shimmon, R. & McDonagh, A.M., 2012. Synthesis and impurity profiling of MDMA
prepared from commonly available starting materials. Forensic science international, 223(1-3),
pp.306–13.
Gimeno, P. et al., 2005. A study of impurities in intermediates and 3,4methylenedioxymethamphetamine (MDMA) samples produced via reductive amination routes.
Forensic science international, 155(2-3), pp.141–57.
Green, A.R. et al., 2012a. Ecstasy cannot be assumed to be 3,4-methylenedioxyamphetamine
(MDMA). British Journal of Pharmacology, 166(5), pp.1521–1522.
Green, A.R. et al., 2012b. Lost in translation: preclinical studies on 3,4methylenedioxymethamphetamine provide information on mechanisms of action, but do not
allow accurate prediction of adverse events in humans. British journal of pharmacology, 166(5),
pp.1523–36.
Green, A.R. et al., 2003. The Pharmacology and Clinical Pharmacology of 3 , 4Methylenedioxymethamphetamine ( MDMA , “ Ecstasy ”). Pharmacological Reviews, 55(3),
pp.463–508.
Holland, J., 2001. ECSTASY:The Complete Guide 1st ed., Rochester, Vermont: Park street press.
Kalant, H., 2001. The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. CMAJ :
Canadian Medical Association journal = journal de l’Association medicale canadienne, 165(7),
pp.917–28.
De la Torre, R. et al., 2004. Human pharmacology of MDMA: pharmacokinetics, metabolism, and
disposition. Therapeutic drug monitoring, 26(2), pp.137–44.
21
De la Torre, R. et al., 2000. Non-linear pharmacokinetics of MDMA (’ecstasy') in humans. British
journal of clinical pharmacology, 49(2), pp.104–9.
Van Laar, M.., 2012. Nationale Drug Monitor jaarbericht 2011, Utrecht.
Liechti, M.E., Gamma, A. & Vollenweider, Franz X, 2001. Gender differences in the subjective effects
of MDMA. Psychopharmacology, 154(2), pp.161–168.
Liechti, M.E. & Vollenweider, F X, 2000. Acute psychological and physiological effects of MDMA
(“Ecstasy”) after haloperidol pretreatment in healthy humans. European
neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology,
10(4), pp.289–95.
McCann, U.D., Eligulashvili, V. & Ricaurte, George A, 2000. (“ Ecstasy ”) -Induced Serotonin
Neurotoxicity : Clinical Studies. Neuropsychobiology, 2000(42), pp.11–16.
Mithoefer, M.C. et al., 2013. Durability of improvement in post-traumatic stress disorder symptoms
and absence of harmful effects or drug dependency after 3,4methylenedioxymethamphetamine-assisted psychotherapy: a prospective long-term follow-up
study. Journal of psychopharmacology (Oxford, England), 27(1), pp.28–39.
Mithoefer, M.C. et al., 2011. The safety and efficacy of {+/-}3,4-methylenedioxymethamphetamineassisted psychotherapy in subjects with chronic, treatment-resistant posttraumatic stress
disorder: the first randomized controlled pilot study. Journal of psychopharmacology (Oxford,
England), 25(4), pp.439–52.
Mohamed, W.M.Y. et al., 2011. MDMA: interactions with other psychoactive drugs. Pharmacology,
biochemistry, and behavior, 99(4), pp.759–74.
Oesterheld, J.R., Armstrong, S.C. & Cozza, K.L., 2004. Ecstasy: pharmacodynamic and
pharmacokinetic interactions. Psychosomatics, 45(1), pp.84–7.
Parrott, A.C., 2001. Human psychopharmacology of Ecstasy (MDMA): a review of 15 years of
empirical research. Human psychopharmacology, 16(8), pp.557–577.
Parrott, A.C., 2012. MDMA and 5-HT neurotoxicity: the empirical evidence for its adverse effects in
humans - no need for translation. British journal of pharmacology, 166(5), pp.1518–20;
discussion 1521–2.
Parrott, A.C., 2007. The psychotherapeutic potential of MDMA (3,4methylenedioxymethamphetamine): an evidence-based review. Psychopharmacology, 191(2),
pp.181–93.
Ricaurte, G A, Yuan, J & Mccann, U D, 2000. 3,4-Methylenedioxymethamphetamine (“ Ecstasy ”) Induced Serotonin Neurotoxicity : Studies in Animals. Neuropsychobiology, 10(42), pp.5–10.
Ricaurte, George A et al., 2002. Severe dopaminergic neurotoxicity in primates after a common
recreational dose regimen of MDMA (“ecstasy”). Science (New York, N.Y.), 297(5590), pp.2260–
3.
22
Rietjens, S.J. et al., 2012. Pharmacokinetics and pharmacodynamics of 3,4methylenedioxymethamphetamine (MDMA): interindividual differences due to polymorphisms
and drug-drug interactions. Critical reviews in toxicology, 42(10), pp.854–76.
Steele, T.D., Mccann, Una D & Ricaurte, George A, 1994. ( MDMA , “ Ecstasy ”): pharmacology and
toxicology in animals and humans. Addiction, (89), pp.539–551.
Sun-edelstein, C., Tepper, S.J. & Shapiro, R.E., 2008. Drug-induced serotonin syndrome : a review.
Expert Opion Drug Safety, 7(5), pp.587–596.
Turner, J.J. & Parrott, A.C., 2000. “Is MDMA a human neurotoxin?”: diverse views from the
discussants. Neuropsychobiology, 42(1), pp.42–8.
Vearrier, D. et al., 2012. Methamphetamine: history, pathophysiology, adverse health effects,
current trends, and hazards associated with the clandestine manufacture of
methamphetamine. Disease-a-month : DM, 58(2), pp.38–89.
23