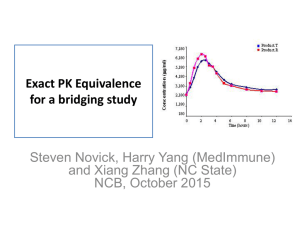
Multisource (Generic) Pharmaceutical Products
advertisement
 Pharmaceutical Products")
MARKETING AUTHORIZATION OF PHARMACEUTICAL PRODUCTS WITH SPECIAL REFERENCE TO MULTISOURCE (GENERIC) PRODUCTS: A MANUAL FOR DRUG REGULATORY AUTHORITIES Annex 3: *Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability 1 Table of Contents of Annex 3 Introduction Glossary Part One. Regulatory assessment of interchangeable multisource Pharmaceutical products 1. General considerations 2. Multisource products and interchangeability 3. Technical data for regulatory assessment 4. Product information and promotion 5. Collaboration between drug regulatory authorities 6. Exchange of evaluation reports Part Two. Equivalence studies needed for marketing authorization 7. Documentation of equivalence for marketing authorization 8. When equivalence studies are not necessary 9. When equivalence studies are necessary and types of studies required In vivo studies In vitro studies Part Three. Tests for equivalence 10. Bioequivalence studies in humans Subjects Design Studies of metabolites Measurements of individual isomers for chiral drug substance products Validation of analytical test methods Sample retention Statistical analysis and acceptance criteria Reporting of results 11. Pharmacodynamic studies 12. Clinical trials 13. In vitro dissolution Part Four. In vitro dissolution tests in product development and quality control Part Five. Clinically important variations in bioavailability leading to non-approval of the product Part Six. Studies needed to support new post-marketing manufacturing conditions Part Seven. Choice of reference product Authors References Appendix 1 Examples of national requirements for in vivo equivalence studies for drugs included in the WHO Model List of Essential Drugs (Canada, Germany and the USA, August 1994). Appendix 2 Explanation of the symbols used in the design of bioequivalence studies in humans, and other commonly used pharmacokinetic abbreviations Appendix 3 Technical aspects of bioequivalence statistics 2 Overview • Why is bioequivalence needed? • What are the ways of demonstrating bioequivalence? • When are bioequivalence studies needed/not needed or may be waived by a regulatory agency? • • • • Design of comparative bioavailability studies Bioequivalence standards (acceptance ranges) Some statistical considerations Other issues - selection of reference product, extended-release delivery systems, stereoisomers • Critical parameters to look into when evaluating dossiers with respect to bioequivalence studies • Some useful reference materials 3 Why is bioequivalence needed? • Pharmaceutical equivalence does not necessarily mean therapeutic equivalence • Multisource drug products should conform to the same standards of quality, safety and efficacy required of the reference product and must be interchangeable Differences in excipients or manufacturing process may lead to differences in product performance. Also, in vitro dissolution does not necessarily reflect in vivo bioavailability. 4 What are the ways of demonstrating therapeutic equivalence? • • • • Comparative bioavailability (bioequivalence) studies Comparative pharmacodynamic studies in humans Comparative clinical trials In vitro dissolution tests 5 Bioequivalence studies are not needed when the multisource product is: a) an aqueous solution for parenteral use b) a solution for oral use c) a gas d) a powder for reconstitution as a solution for oral or parenteral use e) an otic or ophthalmic solution f) a topical aqueous solution g) an inhalation product or nasal spray as an aqueous solution For e, f and g, formulation of multisource product must be similar to reference product. Also, bioequivalence studies may be waived for compositionally similar strengths when one strength in a range has been studied. 6 Bioequivalence studies are particularly needed for pharmaceutical products for systemic action such as: a) Oral immediate release when one or more of the following criteria apply: i) indicated for serious conditions requiring assured therapeutic response ii) narrow therapeutic window/safety margin; steep dose-response curve iii) complicated pharmacokinetics iv) unfavourable physicochemical properties, e.g., low solubility v) documented evidence for bioavailability problems related to the drug vi) where a high ratio of excipients to active ingredients exists b) Non-oral and non-parenteral, such as transdermal patches, suppositories c) Modified release d) Fixed combination 7 Products for non-systemic use Comparative clinical or pharmacodynamic studies are required to prove equivalence for non-solution pharmaceutical products that are for nonsystemic use (oral, nasal, ocular, dermal, rectal, vaginal, etc. application) and are intended to act without systemic absorption. 8 Design of comparative bioavailability studies • Studies should be carried out in accordance with provisions of guidelines on Good Clinical Practice, Good Manufacturing Practice, Good Laboratory Practice • Most common design is single-dose, randomized, two-way crossover study (non-replicated) • Other designs possible, e.g. parallel design for drugs with long half-lives or in patients, steady-state studies for some non-linear drugs 9 Factors to consider in the design of a study • • • • • • • • • Protocol must state a priori, the study objectives and methods to be used Study formulation should be representative of formulation to be marketed Subjects - number - health status - age, weight, height - ethnicity - gender - special characteristics e.g. poor metabolizers - smoking - inclusion/exclusion criteria specified in protocol Randomization Blinding Sampling protocol Washout period Administration of food and beverages during study Recording of adverse events 10 Assay validation • • • • • specificity accuracy precision sensitivity stability • must cover before- and within-study phases • calibration range must be appropriate 11 Bioequivalence standards (acceptance ranges) • The 90% confidence interval of the relative mean AUC of the test to reference product should be between 80-125%. • The 90% confidence interval of the relative mean CMAX of the test to reference product should be between 80-125%. Since CMAX is recognized as being more variable than the AUC ratio, a wider acceptance range may be justifiable. • These standards must be met on log-transformed parameters calculated from the measured data • If the measured potency of the multisource formulation differs by more than 5% from that of the reference product, the parameters may be normalized for potency. • TMAX may be important for some drugs 12 Some statistical considerations • A priori specification of methods Statistical methods to be used must be specified beforehand in the protocol • Number of subjects Take into consideration error variance of parameter, desired significance level and acceptable deviation from reference product Minimum 12 subjects. Usually 18-24 subjects sufficient. • Log-transformation AUC and CMAX should be analyzed after log-transformation Satisfies assumption of Analysis of Variance (ANOVA model is additive rather than multiplicative) • Outliers Must be valid medical reason to drop outlier from analysis 13 Other issues • Selection of a reference product -should be a product for which the safety, efficacy and quality are well established, usually the innovator’s product • Modified-release delivery systems -greater safety concern due to possibility of dose-dumping -may be more difficult to establish equivalence • Stereoisomers -multisource product must have same proportion of enantiomers in the formulation -non-stereospecific assay usually adequate in determination of bioequivalence 14 Critical parameters to look into when evaluating bioequivalence studies • Is the reference product suitable? • Was the study design such that variability due to factors other than the product was reduced? Other design issues e.g. sample size, sampling protocol • Assay validation adequate? • Pharmacokinetic analysis appropriate? • Statistical analysis appropriate? • Acceptance criteria met? 15 Some References • WHO bioequivalence guideline: Marketing Authorization of Pharmaceutical Products with Special Reference to Multisource (Generic) Products. A Manual for a Drug Regulatory Authority (WHO) • Assay validation: Conference report on analytical methods validation: Bioavailability, Bioequivalence and Pharmacokinetic Studies, Pharmaceutical Research Vol.9, No.4, 1992 • Sample size calculation in comparative bioavailability studies: Sample size determination for bioequivalence assessment by means of confidence intervals, Diletti E et al. Int J Clin Pharmacol Ther Toxicol1991, 29:1-8 • Statistical method: A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability, Schuirman DJ, J Pharmacokinet Biopharm, 1987, 15:657-680. • Drug characteristics: American Hospital Formulary Service Drug Information Physician’s Desk Reference • Comprehensive text: Generics and Bioequivalence, Ed. A.J. Jackson, CRC Press, 1994 16 Conclusion • Multisource products should meet the same standards of quality, safety and efficacy as the reference products AND must be interchangeable • Bioequivalence to a reference product demonstrates safety and efficacy of the multisource product and is a good indicator of interchangeability 17