Analytical method development
advertisement

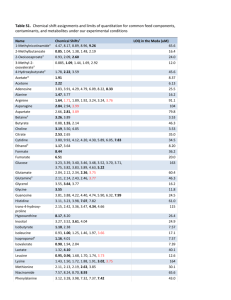
Pharmaceutical Development Training Workshop on Pharmaceutical Development with focus on Paediatric Formulations Protea Hotel Victoria Junction, Waterfront Cape Town, South Africa Date: 16 to 20 April 2007 | Slide 1 of 43 April 2007 Pharmaceutical Development Analytical Method Development Presenter: János Pogány, pharmacist, PhD pogany.janos@chello.hu WHO expert | Slide 2 of 43 April 2007 Analytical Method Development Outline and Objectives of presentation Introduction, guidelines Dossier requirements – Assay – Related substances – Other issues Main points again | Slide 3 of 43 April 2007 Training Workshop on Pharmaceutical Development with focus on Paediatric Formulations Introduction, guidelines Interchangeability (IC) INTERCHANGEABILITY (IC) OF MULTISOURCE FPPs = (ESSENTIAL SIMILARITY WITH INNOVATOR FPP) = PHARMACEUTICAL EQUIVALENCE (PE) + BIOEQUIVALENCE (BE) IC = PE + BE | Slide 5 of 43 April 2007 Pharmaceutical equivalence FPPs meet same or comparable standards (e.g., marketing authorization, analytical methods) – – – – Same API (chemical and physical equivalence) Same dosage form and route of administration Same strength Comparable labeling Pharmaceutical development equivalence Stability equivalence WHO-GMP (manufacturing equivalence) | Slide 6 of 43 April 2007 Prequalification requirements Analytical method validation is required by WHO for the prequalification of product dossiers. Non-compendial ARV APIs and FPPs were/are tested with methods developed by the manufacturer. Analytical methods should be used within GMP and GLP environments, and must be developed using the protocols and acceptance criteria set out in the ICH guidelines Q2(R1) | Slide 7 of 43 April 2007 Guidelines used in PQP „WHO-GMP 4.11 „It is of critical importance that particular attention is paid to the validation of analytical test methods, automated systems and cleaning procedures.” Appendix 4. Analytical method validation (in WHO Expert Committee on Specifications for Pharmaceutical Preparations. 40th Report. Geneva, WHO, 2006 (WHO Technical Report Series, No. 937). http://whqlibdoc.who.int/trs/WHO_TRS_937_eng.pdf Validation of analytical procedures: text and methodology Q2(R1) ICH Harmonized Tripartite Guidelines, (2005) http://www.ich.org/LOB/media/MEDIA417.pdf | Slide 8 of 43 April 2007 General requirements Qualified and calibrated instruments Documented methods Reliable reference standards Qualified analysts Sample selection and integrity Change control | Slide 9 of 43 April 2007 Measure of variation (spread of data) 68.26% 95.46% | Slide 10 of 43 April 2007 Mean (average) chart Abnormal variation of process – special causes USL Upper specification limit average = mean LSL Lower specification limit Abnormal variation of process – special causes | Slide 11 of 43 April 2007 Normal variation due to common causes Capable process Almost all the measurements of a stable process fall inside the specification limits USL – LSL Cp 6 8 10 1.00 1.33 1.66 2.00 OoS results: .27%. 6 ppm 64 ppm http://www.itl.nist.gov/div898/handbook/pmc/section1/pmc16.htm | Slide 12 of 43 April 2007 12 2 ppb NEVIRAPINE – Reference Standard Injection System suitability requirement: RSD is NMT 0.85% | Slide 13 of 43 April 2007 tR A 1 2 3 6.160 6.167 6.166 12466 12311 12432 4 5 6 6.172 6.165 6.168 12530 12457 12479 Mean STD RSD 6.166 0.004 0.06% 12446 74 0.59% Training Workshop on Pharmaceutical Development with focus on Paediatric Formulations Dossier requirements Use of analytical methods - generics Clinical Pharmaceutical Methods At initial phase of pharmaceutical development To determine bioavailability in healthy volunteers To develop a stable and reproducible formulation for the manufacture of bioequivalence, dissolution, stability and pilot-scale validation batches To understand the profile of related substances and to study stability and start measuring the impact of key product and manufacturing process parameters on consistent FPP quality At advanced phase of pharmaceutical development To prove bioequivalence after critical variations to the prequalified dossier | Slide 15 of 43 To optimize, scale-up, and transfer a stable and controlled manufacturing process for the prequalification product April 2007 To be robust, transferable, accurate, and precise for specification setting, stability assessment, and QC release of prequalified batches Analytical procedure characteristics Type of characteristic Identification Impurities Quantitative Assay Limit Accuracy - + - + Precision - + - + Specificity + + + + Detection limit - - + - Quantitation limit - + - - Linearity - + - + Range - + - + Robustness + + + + | Slide 16 of 43 April 2007 Accuracy - ISO 5725 1-6 Accuracy Precision Trueness Systematic errors (Random errors) Intra-assay variability Intra-laboratory variability Repeatability Inter-laboratory variability Intermediate precison Reproducibility Source: ISO. 1994. ISO 5725 1-6: Accuracy (Trueness and Precision) of Measurement Methods and Results. ISO, Geneva, Switzerland. | Slide 17 of 43 April 2007 Accuracy and precision Inaccurate & imprecise Inaccurate and imprecise Inaccurate but precise Accurate but imprecise Precise | Slide 18 of 43 Accurate April 2007 Accurate and precise Percent accuracy (hypothetical figures) Sample LA, % Nevirapine, mg Added Recovered Recovery % RSD % 1 2 50 70 0.499 0.703 0.495 0.701 99.2 99.8 0.64 0.31 3 4 5 80 100 120 0.796 1.001 1.211 0.795 1.005 1.209 99.9 100.4 99.8 0.27 1.88 0.38 6 130 Mean 1.299 0.918 1.296 0.917 99.8 99.8 1.12 0.77 The data show that the recovery of analyte in spiked samples met the evaluation criterion for accuracy (100 ± 2.0% across 50–130% of target concentrations). | Slide 19 of 43 April 2007 % Recovery Percent accuracy (hypothetical figures) 104 103 102 101 100 99 98 97 96 0 1 2 3 4 5 6 Number of samples Red line: LA | Slide 20 of 43 April 2007 Green lines: USL and LSL 7 Precision (of any process) Measured mean Real mean The precision (VARIABILITY) of an analytical procedure is usually expressed as the standard deviation (S), variance (S2), or coefficient of variation (= relative standard deviation, RSD%.) of a series of measurements. The confidence interval (CI) should be reported for each type of precision investigated. PRECISION | Slide 21 of 43 April 2007 Repeatability (of any process) Measured mean REPEATABILITY | Slide 22 of 43 April 2007 Repeatability expresses the precision (spread of the data, variability) under the same operating conditions over a short interval of time. Repeatability is also termed intra-assay precision. Repeatability (hypothetical figures) Injection Peak area Imp1 1 57935 0.301 2 57833 0.301 3 57497 0.299 4 57617 0.300 5 57778 0.301 6 57231 0.298 Mean 57649 0.300 STD 257 0.0013 RSD 0.4% 0.4% 270 0.0014 95% CI | Slide 23 of 43 April 2007 The repeatability precision obtained by one analyst in one laboratory was 1.25% RSD for the analyte and, therefore, meets the evaluation criterion of RSD ≤2%. Intermediate Precision and Reproducibility (of any process) Measured means Intermediate precision expresses within-laboratories variations. #1, #2 and #3: different days, different analysts, different (manufacturing) equipment, etc. Reproducibility expresses the precision between laboratories #1, #2 and #3 (collaborative studies, usually applied to standardization of methodology). (Transfer of technology) Intermediate precision or Reproducibility | Slide 24 of 43 April 2007 Intermediate precision (ruggedness) Sample 1 | Assay (mg/5ml) 51.7 52.6 2 3 4 51.9 53.0 52.5 52.1 52.3 52.9 5 6 Mean 52.3 52.7 52.4 53.2 53.1 52.7 STD RSD 95% CI 0.49 0.9% 0.51 0.44 0.8% 0.46 Slide 25 of 43 April 2007 Combined values Mean STD RSD 95% CI 52.5 0.48 0.9% 0.31 Specificity (selectivity) Specificity is the ability to assess unequivocally the analyte in the presence of components, which may be expected to be present. Typically these might include impurities, degradants and excipients. An example of specificity criterion for an assay method is that the analyte peak will have baseline chromatographic resolution of at least 2.0 minutes from all other sample components Stability indicating analytical methods should always be specific. | Slide 26 of 43 April 2007 Identification – a special case Diethylene glycol (DEG) in paediatric dosage forms has been implicated as the causative agent in numerous deaths since 1937. The victims were mainly children. Illustrative analytical issues of investigation – IR identity test was able to detect DEG at about 20 %w/w – Testing of DEG in Glycerol (and in Propylene Glycol) was recommended with a LOD (sensitivity) of NLT 0.1 %. For detecting DEG at low levels, GC seemed preferable. – The assay was the most relevant test (accurate within ± 0.2%) Illustrative regulatory issues – Legislation – GMP Specificity is an essential but not sufficient characteristic of identification | Slide 27 of 43 April 2007 Specificity (hypothetical figures and data) HPLC chromatograms of (a) API reference standard, (b) FPP and (c) placebo | Slide 28 of 43 April 2007 SPECIFICITY – degradants Stress A (%) * Initial 100.0 Acid 99.3 Peroxide 99.8 All others 100.0 Purity angle 0.040 0.105 0.725 0.045 0.120 1.040 0.060 0.110 0.690 NA Purity threshold 0.280 0.380 1.630 0.280 0.410 1.610 0.270 0.360 1.250 NA There were no peaks in the placebo chromatogram at the retention times of nevirapine (N), methylparaben (MP) and propylparaben (PP) peaks. *Sum of N, MP and PP peak areas. The three ingredients can be assessed in the presence of (nonexpected) degradants. The peaks are homogeneous and pure. The method is selective, specific and stability-indicating. | Slide 29 of 43 April 2007 LOD, LOQ and SNR Limit of Quantitation (LOQ) Limit of Detection (LOD) Signal to Noise Ratio (SNR) Peak A LOD Baseline | Slide 30 of 43 noise April 2007 Peak B LOQ LOD and LOQ (hypothetical figures) Impurity 1 Injection | LOD Impurity 2 LOQ LOD LOQ 1 4176 7235 3497 7892 2 3608 8099 4258 7791 3 4196 7950 3275 8292 4 4303 8166 3464 8050 5 3932 7847 4008 8368 6 5238 8415 4702 8284 Mean 4242 7952 3867 8113 STD 548 402 551 238 RSD 12.9% 5.1% 14.3% 2.9% Conc. (μg/ml) 0.086 0.171 0.107 0.214 Conc. (%w/w) 0.017 0.033 0.019 0.039 Slide 31 of 43 April 2007 LOD and LOQ The limit of detection (LOD) is defined as the lowest concentration of an analyte in a sample that can be detected, not quantified. It is expressed as a concentration at a specified signal : noise ratio (SNR), usually between 3 and 2 : 1. In this study, the LOD was determined to be 0.086 μg/ml (Impurity 1) with a signal : noise ratio of 3.6 : 1 The limit of quantitation (LOQ) is defined as the lowest concentration of an analyte in a sample that can be determined with acceptable precision and accuracy under the stated operational conditions of the method. The ICH has recommended a signal : noise ratio (SNR) of 10:1. The LOQ was 0.171 μg/ml (Impurity 1) with a signal:noise ratio of 11.3. The RSD for six injections of the LOQ solution was ≤2%. | Slide 32 of 43 April 2007 Linearity Measured mean | Slide 33 of 43 Real mean April 2007 Precision Linearity expresses differences in precision at different points of a given range. „The linearity of an analytical procedure is its ability (within a given range) to obtain test results, which are directly proportional to the concentration (amount) of analyte in the sample.” Linearity and range 5000 y = 36.124x - 7.2984 Assay mean 4000 2 R = 0.9998 3000 2000 1000 0 0 20 40 60 80 100 120 140 Concentration, % Acceptance criterion: correlation coefficient should not be less than 0.9990 | Slide 34 of 43 April 2007 Linearity and range Concentration range 1.0–1.3 mg/ml (10–130% of the theoretical concentration in the test preparation, n=3) Regression equation was found by plotting the means of peak area (y) against the analyte concentration (x) expressed in %: y = 36.124x - 7.2984 (R2 = 0.9998). The regression coefficient demonstrates an excellent relationship between peak area and concentration of analyte. The analyte response is linear across 10-130% of the target nevirapine concentration. | Slide 35 of 43 April 2007 Range (minimum requirements) Assay of an API or a FPP: ± 20% of the test concentration. Content uniformity: ± 30% of the test concentration (unless a wider more appropriate range, based on the nature of the dosage form (e.g., metered dose inhalers), is justified). Dissolution testing: ± 20 % over the specified range. Impurity: from the reporting level of an impurity to 120% of the specification. (Unusually potent or toxic impurities, LOD and LOQ should be commensurate with ICH requirement.) If assay and purity are performed together as one test and only a 100% standard is used, linearity should cover the range from the reporting level of the impurities to 120% of the assay specification | Slide 36 of 43 April 2007 Stability of analytical solution Stability Measured means Stability (of the analytical solution) expresses variation of the measured mean as a function of time. #1 … First measurements #2, #3, #4, …n Series of measurements of the same sample within a relatively short period of time. | Slide 37 of 43 April 2007 Stability of test analytical solution Time in hours | Impurity-1 Area Difference 0 72079 1 71574 0.7% 2 71740 0.5% 3 71960 0.2% 4 72352 -0.4% 5 71573 0.7% 10 72322 -0.3% 15 72310 20 72312 25 72670 Slide 38 of 43 April 2007 An analytical solution prepared from Nevirapine 50mg/5ml Oral Suspension was spiked with Impurity-1 at specification level and stored in a capped volumetric flask on a laboratory bench at uncontrolled room temperature under normal lighting conditions for 25 hours. Conclusion: the stability of the -0.3% analytical solution of Impurity-1 is -0.3% not a source of variation. -0.8% Sensitivity and robustness | Slide 39 of 43 April 2007 Robustness Method parameter STP Flow Wavelength Variation of mobile phase Column temperature pH | Slide 40 of 43 April 2007 Variation -10% 10% -5nm +5nm -2% +2% -5oC +5oC -0.3 +0.3 tR Impurity 1 0.83 0.83 0.84 0.82 0.83 0.80 0.84 0.82 0.83 0.83 0.83 Impurity 2 1.80 1.81 1.82 1.81 1.81 1.89 1.76 1.80 1.81 1.81 1.80 Methods for cleaning validation Method for assay and related substances used in stability studies of API and FPP – Specificity (in samples taken from a cleaning assessment) – Linearity of response (from 50% of the cleaning limit to 10x this concentration; R2 ≥ 0.9900; ) – Precision • Repeatability (RSD ≤ 5%) , • intermediate precision [ruggedness (USP)], and • reproducibility – Limits of detection and quantitation – Accuracy or recovery from rinsate (≥ 80%), swabs (≥ 90%), and process surface (≥ 70%) – Range (lowest level is at least 2x higher than LOQ) | Slide 41 of 43 April 2007 Main Points Again Analytical procedures play a critical role in pharmaceutical equivalence and risk assessment / management: – establishment of product-specific acceptance criteria, and – stability of APIs and FPPs. Validation should demonstrate that the analytical procedure is suitable for its intented purpose. HPLC systems and method validation deserves special attention during the assessment of dossiers for prequalification. | Slide 42 of 43 April 2007 THANK YOU | Slide 43 of 43 April 2007