bioprecipitates14
advertisement

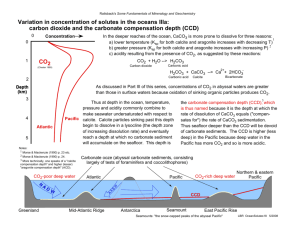
Proterozoic Stromatolites Nastopoka Sound Carbonates et al Francis, 2014 Bio-Chemical Sediments Cations that are leached during weathering and transported by solution in water include: K+, Na+, Ca++, Mg++ Other cations are also present in solution, but at very low concentrations: Si4+, Mn2+, Fe2+ River Input versus Sea Water Sink Species Av. Rivers (ppm) HCO3SO4-Cl- 50 10 8 140 2,715 18,980 Ca++ Mg++ Na+ K+ 15 4.1 6.3 2.3 413 1,290 10,800 399 0.04 6.5 0.00006 2.8 Fe Si Ocean (ppm) River Input versus Sea Water Sink Species Av. Rivers (ppm) Ocean (ppm) HCO3SO4-Cl- 50 10 8 140 2,715 18,980 Ca++ Mg++ Na+ K+ 15 4.1 6.3 2.3 413 1,290 10,800 399 0.04 6.5 0.00006 2.8 Fe Si Solubility of Minerals in Sea Water at 25oC Mineral Formula Solubility (ppm) Quartz Opal SiO2 SiO2.nH2O Calcite Aragonite Dolomite CaCO3 CaCO3 CaMg(CO3)2 14 15 32 Gypsum Anhydrite CaSO4.2H2O CaSO4 2,400 2,500 1,400 Halite Sylvite Carnalite NaCl KCl KMgCl3.6H2O 360,000 350,000 650,000 30,000 10 Conc (ppm) 1 120 120 Calcite Group 32/m Rhombohedral Although calcite (s.g. 2.75) is the stable form of CaCO3 at room pressure and temperature and is supersaturated in sea water, it has never been successfully precipitated from seawater in experiment. Some organism (eg. echinoderms) secrete metastable high-Mg calcite with 4 to 30% dissolved MgCO3, but this is converted to calcite during diagensis. In past epochs, high-Mg calcite has been the dominant biochemical precipitate in the oceans, rather than aragonite, which is the dominate carbonate precipitated in the oceans today. Dolomite Group 3 Carbonates: Three groups of carbonate minerals: Rhombohedral Although dolomite (s.g. = 2.85) is a common rock in the stratigraphic record, dolomitic sediments are rare in the modern environment, and, despite the fact that seawater is supersaturated in dolomite, it too has never been directly precipitated from seawater in experiment. Aragonite Group 2/m2/m2/m Orthorhombic Aragonite (s.g. 3.94) is the high pressure polymorph of CaCO3 and is slightly more soluble in sea water than calcite. Aragonite, however, is the dominant form of CaCO3 that precipitates directly from seawater today, and is the dominant skeletal material of most carbonate-secreting organisms. Recrystallization of aragonitic sediments in contact with fresh water, converts aragonite to calcite, in much the same way that amorphous silica is converted to quartz. Controls on the Solubility of Calcite: CaCO3 + CO2 + H2O = Ca2+ + 2HCO3K= [Ca2+] [HCO3-]2 [CaCO3] [CO2] [ H2O] = CO2 + H2O = [Ca2+] [HCO3-]2 [CO2] [ H2O] in presence of calcite H2CO3 (carbonic acid) H2CO3 + CaCO3 = Ca2+ + 2HCO3- (bicarbonate ion) The concentration of Ca2+ in water in equilibrium with calcite and air (PCO2 = 3 × 10-6 atm) is approximately 20 ppm, which corresponds to a calcite solubility of approximately 50 ppm. the Solubility of Calcite: CaCO3 + CO2 + H2O = Ca2+ + 2HCO3K= [Ca2+] [HCO3-]2 [CaCO3] [CO2] [ H2O] Inspection of the above equilibria indicates that the solubility of calcite is directly related to the amount of dissolved CO2, which is an inverse function of temperature. Thus the higher the temperature, the lower the solubility of calcite. On the other hand, increased pressure puts more CO2 into a solution and increases the solubility of CaCO3. The combined effects of increasing pressure and decreasing temperature in the oceans are such that stable carbonate sediments are restricted to water depths of less than 3500 metres. Below this “carbonate compensation depth” there is a dramatic increase in the solubility of CaCO3, such that it becomes undersaturated in seawater, and carbonate sediments are rare because they dissolve. Note that the greater solubility of aragonite compared to calcite means that aragonite becomes undersaturated in seawater at much shallower depths. This results in the conversion of aragonite to calcite during diagenesis. The above relationships are complicated by the fact that the solubility of a given mineral is sensitive to the other species in solution, which compete and/or interfere with the species of interest. Effectively, the presence of other ions reduces the activity coefficient of the ions of interest in the equation for the equilibrium constant, thus requiring higher concentrations to achieve saturation. The magnitude of this effect is proportional to the ionic strength of the solution: I = ½ × Mi × Zi2 where Mi are the molar concentrations of the species and Zi are their ionic charges. Note: neutral species are assumed to have no effect. Furthermore, the kinetics of nucleation play as large a role as solubility and concentration in determining the point at which a mineral will precipitate from solution. For example, although calcite will precipitate from fresh water, it will not from seawater, despite the fact that seawater is supersaturated in CaCO3. This may reflect the fact that the high concentration of Mg(OH)2 in sea water poisons the nucleation and growth of calcite. In general, many dissolution reactions seem to be one way, that is it is much easier to dissolve a mineral than to precipitate it. And then there is life, which plays havoc with everything. Ostwald’s Step Rule In general, the least stable polymorph of a chemical compound tends to be the first to precipitate from a solution. Thus aragonite precipitates before calcite, opal precipitates before quartz. In modern oceans, the relatively unstable polymorph aragonite is precipitated by organisms rather than calcite, despite the fact that it has a higher solubility. Some have argued than in fact CaCO3 precipitates first as a amorphous gel that is converted rapidly to aragonite, and then later to calcite during diagenesis. Allochthonous Limestones Most limestones were originally clastic rocks, and in many respects can be classified according to the same grain size criteria as the siliciclastic rocks: mudstones, shale lutite, micrite sandstones grainstone conglomerates & breccia rudite These types of limestone are referred to as allochthonous, in that their constituents have been transported from elsewhere. The major differences between allochthonous limestones and siliciclastic rocks are in the nature of the clastic fragments, the degree of sorting, and the matrix or cement that holds these rocks together. Allochthonous versus Autochthonous Carbonates Reef complexes are essentially living buckets, in which the walls of the bucket are composed of boundstone of living corals and related species, the shallow inner lagoon is filled with fine grainstones and mudstone, while the steep outer flanks are composed of coarser grainstones and rudestone, formed from talus off the reef front. Boundstones Stromatolites and Algal Mats Mats of blue green algae that grow in the intertidal to supratidal zone. These organic mats trap carbonate mud when flooded, eventually forming a finely laminated micrite, in which the original organic seams have been replaced by porous septa or sparite cement. One of the few clastic sedimentary rocks in which the size of porosity is larger than the grains. Dominant carbonate rock in the Precambrian, before the development of reefs. Stromatolites represent localized buildups or cabbage-shaped heads formed by a similar mechanism. Distribution of Carbonate Sediments Depth: Active growing reefs are restricted to the photic zone in the oceans, which extends only to depths of 20 to 150 m, depending of the turbidity of the water. Clastic carbonates sediments can be transported to much deeper environments, but become unstable at depths greater than 3500 meters because of the high solubility of CaCO3 in cold water at high pressure. All this means that carbonate sedimentation is largely restricted to continental shelfs environments. Reefs will not grow under turbid conditions where there is a high terrigenous clastic input, such as at the mouths of large rivers like the Amazon and the Mississippi. Distribution of Carbonate Sediments Latitude: Reefs grow only within 30o of the equator, restricted to waters with minimum annual temperatures of 18oC. Carbonate clastic sediments, however, can reach 60o on either side of the equator. Apparently, cool water carbonates do form on some high latitude continental shelves. Longitude: Distribution of Carbonate Sediments Carbonate reefs are better developed on the western sides of the Earth's oceans because of the direction of the prevalent wind and ocean currents within 30o of the equator. Deep ocean water tends to rise on the eastern sides of the oceans and sink on the western sides. Because cold water dissolves more CaCO3, the deep cold waters are nutrient-rich, becoming supersaturated as they warm rising to the ocean surface, promoting high planktonic productivity, which in turn results in turbid water conditions. Corals, on the other hand, thrive best in clear lowproductivity ocean water and as a result grow best on the western sides of ocean basins; reaching latitudes of 30o on the western sides of oceans, but only about 5-10o latitude on the east. Carbonate Reef Distribution in Time Precambrian limestones are dominated by stromatolite mats and mounds that develop by the repeated coating of tidal-zone algal mats by micrite. The first true coral reefs developed in the mid-Ordovician and were dominated by HiMg calcite secreting organisms that reached their maximum development at the end of the Devonian. Carbonate development at this point retreated to build ups and mounds formed of aragonite secreting organisms, and then was arrested completely by the mass extinction the end of the Permian. Aragonite reefs became widespread again in the Triassic and Jurassic, but then decreased in the early Cretaceous, with carbonate growth returning to mounds of calcite secreting organisms. Extensive aragonite reefs only returned in the Tertiary and have continued until today, although there now appears to be a reef die-off occurring, possibly as a result of global warming. Dolomite Although dolomite is a common rock in the stratigraphic record, particularly in the Paleozoic where it is the dominate carbonate rock, the mineral dolomite has never been successfully precipitated from sea water solutions at temperatures less than 100oC, possibly because of the strong hydrolysis of Mg (Mg(OH)2 ) in water solutions. Furthermore, modern day dolomitic sediments are relatively rare and largely restricted to sabkha environments (salt-encrusted supra-tidal flats) where they appear to form by the alteration of initial aragonite precipitates. Most dolomites are thought to have formed either early by the penecontemporaneous alteration of unconsolidated calcareous sediments or somewhat later during the diagenesis of carbonate rocks. Four models for penecontemporaneous dolomite: Sabkha or hypersaline model Rapid evaporation in hot dry climates, such as that of the Persian Gulf, increases the salinity of sea water in supra-tidal flats to the point at which gypsum precipitates, which depletes the remaining brines in Ca, raising the Mg/Ca ratio from 5:1 to 10:1. These Mg-rich brines are denser than seawater and sink into the underlying calcareous sediments, reacting with them to convert them to dolomite. An analogous process on a larger scale may be responsible for the stratigraphic association of dolomite with evaporite deposits Mixing zone model It has been proposed that dolomite might form in the mixing zone between ocean water and fresh water. Although the mixing of two fluids can result in the supersaturation of mineral phases, this has yet to be substantiated for the mineral dolomite, and many conditions of mixing actually result in undersaturation. These results reflect the fact that while concentrations of species are linear functions of mixing, the solubility products of minerals and carbonate solubility as a function of PCO2 are non linear. Low-sulfate model The presence of SO42- is known experimentally to inhibit the nucleation of dolomite and dolomite has been precipitated successfully in the absence of SO42-. Thus processes that lower SO42- content have been proposed for the direct precipitation of dolomite from sea water solutions in unconsolidated sediments. Such process might include the bacterial conversion of SO42- to sulfide in anoxic environments, as well as the precipitation of gypsum (CaSO4.2H2O) in the sabkha environment described above. Large through-put model It has been proposed that a large through-put of sea water will, at high water/sediment ratios, convert unconsolidated sediments from calcite, or high-Mg calcite, to dolomite by ion exchange of Mg2+ for Ca2+. Such models require high water/rock ratios. For example, ~ 800 m3 of seawater is required to convert 1m3 of calcite to dolomite. This number drops to ~ 45 m3, if the solution is a brine which has been evaporated to the point of halite saturation. Diagenetic Models for the origin of Dolomite: High Magnesian Calcite Todays carbonate organisms are dominantly aragonitic, but it appears that in the past there have been episodes during which high-Mg calcite was favoured, for reasons which remain somewhat unclear, but likely reflect low Mg/Ca ratios. Hi-Mg calcite appears to be thermodynamically unstable, so that during diagenesis it would revert to calcite or aragonite, giving Mg2+ to solutions that then go on to “dolomitize” other carbonates along the fluid pathways. Evaporite Association Dolomite is commonly associated with evaporite deposits. Following the precipitation of gypsum in evaporite sequences, the Mg/Ca ratios of the residual brines rise rapidly. These residual brines are denser and sink into underlying carbonate, gradually converting it to dolomite. the Solubility of Calcite: CaCO3 + CO2 + H2O = Ca2+ + 2HCO3K= [Ca2+] [HCO3-]2 [CaCO3] [CO2] [ H2O] Temperature and PCO2 Aragonite Calcite aragonite Silica: The solubility of amorphous silica is controlled by a number of possible reactions. At low pH, the solubility is controlled by: SiO2 + 2H2O H4SiO4 (silicic acid) (1) K = [H4SiO4] [SiO2][H2O]2 This reaction is insensitive to pH, however, at high pH ( pH > 8), H 4SiO4, a weak acid begins to dissociate and a significant proportion of dissolved silica is present in the form H 3SiO4H4SiO4 H+ + H3SiO4- K = [H]+ [H3SiO4-] [H4SiO4] K = [H]+ [H3SiO4-] [SiO2][H2O]2 Combining the above two reactions, we get: SiO2 + 2H2O H+ + H3SiO4- (2) = Under these conditions, the solubility of silica a strong function of pH and temperature. Thus, unlike CaCO3, SiO2 solubility is enhanced by higher temperatures and pH’s, but relatively insensitive to pressure. Note: The higher solubility of amorphous silica versus that of crystalline quartz results in the conversion of primary opal to quartz during diagenesis. A similar phenomena results in the conversion of aragonite to calcite during diagenesis. [H]+ [H3SiO4-] in the presence [H2O]2 of amorphous SiO2 Chert River waters are saturated in SiO2 and yet the ocean is undersaturated. Clearly there must be a mechanism for removing silica from ocean water. In the modern environment diatoms (single cell aquatic algae with exterior silica shells) and radiolaria (single cell planktonic protozoan with internal silica tests) construct their skeletons of opaline silica. When these floating pelagic plants and animals die, their skeletons rain to the bottom where they accumulate as a siliceous ooze. Modern oozes are dominated by diatoms, but radiolaria dominated from the early Paleozoic until the Cretaceous. diatoms Without their organic cover, these skeletons start to dissolve forming the silica-saturated interstitial water of the ooze, which then aids the recrystallization of the opal into crytocrystalline quartz during diagenesis. Some cherts exhibit ripple cross lamination indicative of current reworking, but most lack internal structures and commonly all trace of the former organic skeletons are erased during diagenesis. Radiolaria Chert Bedded: Characterized by layers of chert several centimeters in thickness separated by thin seams of siliceous shale. Form in areas where there is little other siliciclastic or carbonate sedimentation, such as deep water marine environments, below the carbonate compensation level. Commonly associated with pillowed volcanic rocks, turbidite sandstones, and other deep water sediments. Nodular: Chert nodules commonly form in shelf-type carbonate rocks during diagenesis due to the remobilization of silica from the skeletons of diatoms and/or radiolarian, as well as sponge spicules. Abyssal Basins (> 2000 m): The deep ocean basins are characterized by water depths of 2000 - 5000 m. The sediments in such environments are typically clays and oozes formed by the accumulation of the skeletons of pelagic organisms that have settled through the water column. Distal turbidites may also be present. Clay and pelagic material also settle out in shallow water environments, but are swamped by the input of clastic sediment. Sedimentation rates in the deep oceans, however, are very low (on the order of 1 cm/ 1000yrs). Red Terrigenous Clay: The most widespread sediment on the abyssal floor is red clay. This clay probably has a complex origin, including wind blown dust, meteoritic dust, volcanic ash, and possible clay sized terrigenous material. The red colour of this sediment probably reflects the relatively oxidizing nature of the cold abyssal waters, which are poor in organic material. Calcareous Oozes: At water depths less than 3500 meters, calcareous oozes are common. These represent the accumulated skeletons of the floating foraminifera, such as "Globigerina", one of the few modern organisms that secrete calcite tests. Such sediments are not found in deeper water, below the carbonate compensation depth, because CaCO3 is undersaturated in these colder deep waters, and the skeletons dissolve. Siliceous Oozes: Below 3500 meters, siliceous oozes become common, formed by the settling of the silica skeletons of diatoms (aquatic algae) and radiolarian (planktonic protozoan). Diatoms occur preferentially in the colder waters of high latitudes. Iron Formation Iron formations are Fe - rich deposits characterized by the cyclic alternation of Fe-rich and silica-rich bands on the scale of centimeters to meters. Although a few Fe-formations are Paleozoic, the majority are Precambrian in age. This age distribution might reflect a lower oxygen fugacity for the Precambrian atmosphere that kept Fe in its more soluble Fe2+ state. Iron Formation Algoma-type – typically Archean - pre 2600 Ma Exhalative deposits typically associated with active volcanic vents in deep marine environments. Associated with mafic and felsic subaqueous volcanics, greywackes, and black shales. Typically relatively restricted in area (a few kms) and thickness (0.1 10 m). Algoma-type Fe-formations have important connections with volcanogenic massive sulfide deposits and commonly provide very useful stratigraphic markers in otherwise monotonous sequences of submarine lavas. Modern “Black Smoker” Porpoise Cove Fe-Formation – 4.0 Ga Iron Formation Consist of finely laminated alternating iron and silica-rich bands. The silica is usually in the form of grey chert or jasper, although in metamorphosed deposits it is commonly converted to more coarsely crystalline quartz. Four distinct facies according to the type of Fe-bearing minerals: sulfide pyrite FeS2 carbonate siderite ankerite FeCO3 & CaFe(CO3)2 silicate greenalite minnesotaite stilpnomelane chamosite Fe3Si2O5(OH)4 Fe3Si4O10(OH)2 Fe-chlorite Fe-chlorite oxide hematite magnetite Fe2O3 & Fe3O4 Fe formations are commonly zoned, with central sulfide and/or carbonate facies closest to volcanic vents and marginal silicate and/or oxide facies furthest from the vent. Oxide facies dominates, but all facies are commonly found in close spatial association. Algoma-type Fe formations are thought to have formed in anoxic hot-spring environments, perhaps by the aid of bacteria. Deposits associated with Black Smokers found along mid-ocean ridges are thought to be modern equivalents Rise in Atmospheric Oxygen Phanerozoic Archean Hadean Proterozoic Superior-type - Proterozoic – 1800 - 2400 Ma Classic banded iron formation (BIF) consists of alternating Ferich and silica-rich bands, with the oxide facies dominating and the sulfide facies rare. Formed on shallow shelves of stable continental margins with little obvious connection to volcanism, for example in the Labrador Trough. BIF are typically one of the first sediments to be deposited in Proterozoic basins, and are commonly associated with coarse clastics and limestones. The aerial extent and thickness (several metres to 1000 m) are typically much greater than Algoma type. Superior-type Fe formations are thought to be formed by the rise of reduced Fe-rich waters from the deep ocean basins onto shallow marine shelves, whose relatively oxidizing environment caused the precipitation of Fe as Fe3+. Phanerozoic Ironstones Oolitic or coated-grain Phanerozoic clastic rocks composed of goethite, hematite, and chamosite coating detrital quartz grains. Typically relatively restricted in terms of area and thickness (1 to 10’s of metres). Formed on shallow marine shelves, principally during the lower Paleozoic and the Jurassic-Cretaceous. The absence of true Fe-formation in the Phanerozoic may reflect the low solubility of Fe3+ in relatively oxidized marine waters. Evaporites Evaporites form in hot dry climates in which evaporation is strong. Modern evaporite deposits are restricted to latitudes around 30o N and 30o S, as are modern deserts, where atmospheric convection patterns result in the downward movement of cold air, which becomes undersaturated in water as it is heated and compressed. 30oN 30oS Marine Evaporites All marine evaporite successions exhibit the same sequence of mineral precipitation because the composition of sea water is relatively constant, and appears to have relatively little changed since the beginning of the Paleozoic. The sequence is: Mineral % total solute % Res ρ Brine aragonite gypsum/anhydrite 0.3 % 4.5 % 50 % 20 % 1.1 1.126 halite sylvite, carnalite, Mg & K salts 77.4 % 17.7 % 10 % <5% 1.214 1.29 Anhydrite forms by the diagenetic dehydration of gypsum with a 38 % volume decrease. There are many back-reactions with the evolving brines, and numerous diagenetic reactions that produce a complex variety of final Mg and K salts. Formation of Marine Evaporites 1000 metres of sea water will produce approximately 16 metres of precipitated evaporite sediment. Clearly the thick ( 1 km) evaporite successions of the interglacial periods of the Cambrian, Devonian, Permian, Cretaceous, and Miocene require mechanism for the processing of enormous volumes of water. Formation of Marine Evaporites Castile Formation Texas Furthermore, many evaporite sequences consist of only one or two dominate facies, which alternate or constitute thick successions, for example: calcite and gypsum/anhydrite, anhydrite and NaCl, or NaCl and KCl. Some recharge mechanism is needed to buffer brine compositions within limited salinity ranges for long periods of time. alternating varves of calcite and gypsum Restricted Basin Recharge Caspian Sea Kora-Bogaz-Gol (Black Throat Lake) Caspian Sea, Turkmenistan 1995 1985 Caspian Sea 1.2% salinity Caspian Sea Kara-Bogaz 35% salinity Three Models for Development of Marine Layered Evaporite Sequences In Restricted Basins: Deep Water - Deep Basin Paleo-Mediterranean Sea (pre-Eocene) Shallow Water - Shallow Basin Persian Gulf Sabkhas Shallow Water - Deep Basin Dead Sea Mediterranean Sea Mediterranean Sea Persian Gulf sabkha Gibraltor Dead Sea Salt Domes off Brazil salt domes salt dome salt dome Continental Evaporites The mineralogy of evaporite sequences developed in continental playas are more varied because the composition of the brine solutions reflects the local environment. The sequence of minerals precipitated becomes critically dependent on the ratios of anion and cation species because of the branching that occurs as each evaporite mineral precipitates. alluvial fans Death Valley Playa Death Valley Salts The solubility of a mineral, and thus the point at which it precipitates from an evaporating brine solution is determined by the product of the activities of its constituent ions in solution: Solubility Product K = (aNa+)2 × (aSO4=) (aNa2SO4) = (XNa+)2 × (XSO4=) 1 Once the solubility product of a mineral is reached, its precipitation will quantitatively remove the lessor ion from solution. The concentration of the more abundant ion, however, will continue to increase with further evaporation. Each saturation point thus represents a junction at which small differences in concentration lead to completing different precipitation sequences. The buffered composition of sea water leads to a constant evaporation sequence in marine exaporites. Small differences in water composition in continental environments, however can lead to distinctly different evaporation sequences, which we take advantage of economically.