chapter 8
advertisement

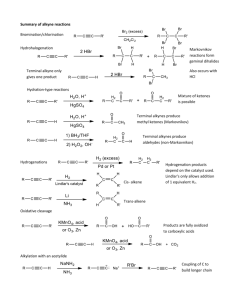
CHEM 3013 ORGANIC CHEMISTRY I LECTURE NOTES CHAPTER 8 1. The Alkyne Functional Group Alkynes are compounds with carbon-carbon triple bonds. Recall that alkanes use sp3 hybrid orbitals and that alkenes use sp2 hybrid orbitals. The alkynes form bonds using a third type of hybrid orbital, the sp orbital. The consequence of this kind of bonding are two fold: (1) Because of the extra "electron glue" provided by the two sets of πelectrons, the alkyne bond is the shortest C-C bond known (about 1.2Å, as compared with 1.33Å for alkenes and 1.54Å for alkanes). and (2) Alkynes are incapable of Cis/Trans geometric isomerism because of the resulting linear configuration of the substituents. H C C H Acetylene (an alkyne) E 2p N E R G Y 2p mix 2s and 2p orbitals 2sp 2s 1s 1s Elemental Carbon sp Hybridized Carbon in Alkynes π-bonds (two 2p orbitals on carbon H H σ-bond (1s orbital on hydrogen and sp orbital on carbon) σ-bond (two sp orbitals on the carbons Bonding in Alkenes 2. Nomenclature of Alkynes The simplest possible alkyne is C2H2 is known by the common name of acetylene. Another name for alkynes as a class is acetylenes. a. IUPAC Nomenclature The systematic nomenclature of alkynes is similar to alkenes. 1 2 1. Name the longest chain containing the triple bond 2. The suffix -ane is replaced by -yne and the triple bond is given the lowest possible number. 3. When double and triple bonds are present in the same molecule, the principal chain is the carbon chain containing the greatest number of double and triple bonds. 4. The compound still ends with the suffix -yne 5. Precedence is given to the naming scheme which gives the lowest number for the first multiple bond regardless whether it is double or triple. 6. In naming the compounds, the double bond is named first...dropping the last e of the -ene suffix. C C C C The eight membered ring is the smallest stable cyclic alkene 2,6-dimethyl-hept-2-yne 3,6,8-trimethyl-4-isopropylcyclooctyne C C C C trans- 5-nonen-3-yne (Z)-3-ethyl-2-nonen-6-yne Compounds that contain both double and triple bonds are named as alkynes. Precedence is given to the naming scheme which gives the lowest number for the first multiple bond regardless if it is a double or triple. In naming, the double bond is cited first. If both double and triple bonds are at equivalent the double bond takes precedence. Alkyne Nomenclature b. Alkynes as Substituents Side-chain groups containing a triple bond are called alkynyl groups and are named by replacing the final -e and adding the suffix -yl.. The alkynyl group is numbered outward from the point of attachment to the principal chain. 4' H 6 4 5 H3C 3' 2' 3 1 2 C CH2 CH3 C Named as a substituent; Fewer carbons than ring 1' H trans-3-(2-butynl)-6-methyl-cyclohexene Naming Alkynes as substituent groups c. Properties of Alkynes Terminal alkynes (those that have a C-H bond) are less stable than internal alkynes. The situation is similar to alkenes (recall that alkene stability increases with alkyl substitution), and for the same reason: Hyperconjugation. Overlap of π-type orbitals with SP3 Hybridized orbitals on adjacent alkyl groups leads to a stabilizing interaction. Structural Isomers H C C CH2CH2CH3 H3C C C CH2CH3 2-Pentyne Internal Alkyne 1-Pentyne Terminal Alkyne E N Heats of Hydrogenation E R H C C CH2CH2CH3 + 2 H2 H3C CH2CH2CH2CH3 G ∆H˚ = -44.2 Kcal/mole Y H3C C C CH2CH3 + 2 H2 H3C CH2CH2CH2CH3 ∆H˚ = -40.9 Kcal/mole 1-pentyne 2-pentyne 3.3 Kcal mole pentane Internal alkynes are more stable than terminal alkynes because of Hyperconjugation. H H C C C H H Energy Differences in Alkynes 3. Synthesis of Alkynes There are relatively few general methods of alkyne syntheses, the best two are: (1) alkylation of acetylene (to be discussed somewhat later), and (2) Twofold elimination of HX from dihalides. Acetylene itself can be made by a simple process of hydrolysis of calcium carbide as described by the following reaction: CaC2 + 2 H 2O → H-C≡C-H + Ca(OH)2. Acetylene is a gas, and was one of the first illuminating gases used in gas lights. The reaction described above is also the source of acetylene used in "carbide cannons" which are often fired off when a touchdown is scored during a football game. A small amount of CaC2 is added to water, acetylene is generated and then ignited in an enclosed space. Thus creating the BOOM. Recall that the elimination of HX from an alkyl halide (upon treatment with a base) will result in the formation of alkenes. The π-bond systems of alkynes can be prepared in much the same manner. Since an alkyne has two π-bonds however, we have to eliminate two molecules of HX. This is usually done by treatment of a vicinal dihalide with an excess of strong base. Vicinal dihalides are the starting material of choice because they are readily available by the addition of bromine or chlorine to an alkene. Thus the overall method of halogenation-dehalogentaion provides an excellent route for going from an alkene to an alkyne. Although different bases can be used for this double elimination of 2HX from dihalides, salts of the amide anion (NH2-) are usually preferred because it's strongly basic nature gives higher yields of products. The twofold elimination process occurs in two discrete steps, passing through a vinyl halide intermediate. 3 4 Alkynes via gem-dihalides H3C CH3 2 eq. strong base C Cl Cl H3C C C H Alkynes via vicinal dihalides H H3C Cl C CH3 2 eq. strong base C H H3C C C CH3 Cl NH2- = Amide anion....conjugate base of ammonia (NH3) pKa (NH3) = 35 Mechanism H H H3C Cl C H C CH3 + NH2 H3C Cl C C CH3 + NH2 Cl Alkynes are formed via two consecutive elimination reactions. H3C C C CH3 Alkynes via Elimination Reactions of Dihalides 4. Electrophilic Reactions of Alkynes a. Addition of HX and X2 The chemistry of alkynes is dominated by electrophilic addition reactions similar to that of alkenes. For example, HX can be added to alkynes to give vinylic halides as products. However, HX will also react with the alkene product ( alkynes are somewhat less reactive than alkenes to electrophilic addition) to result in formation of a geminal dihalide (both halogens on the same carbon). Thus every addition reactions to alkynes will result in some formation of dihalide. The reaction leading to vinyl halide can be maximized by avoiding an excess of HX . The initial addition process follows Markovnikov's rule where applicable. That is, when terminal alkynes react, the vinyl halide produced will be the one in which the H+ (electrophile) has added to the terminal carbon . The reason for this alkyne regioselectivity is directly analogous to that described for alkenes. The electrophile will add so as to form the more stable vinyl cation intermediate. As for simple carbocations, the vinyl cation with the greater amount of alkyl substitution at the carbon bearing the + charge will be more stabilized through hyperconjugation. While regioselectivity is not observed in electrophilic addition to internal alkynes ( the two possible vinyl cations in this case are of equal stability), the reaction often shows stereoselectivity. The vinyl halide produced by addition of HX to an internal alkyne very often (but not always) shows a trans relationship of the H and X. The electrophilic addition of X2 to alkynes leads to vinylic dihalides (alkenes with halogens on each carbon of the double bond). The stereoselectivity of this reaction again leads to trans products. HC CCH2CH2CH3 HCl CH3COOH H2C CClCH2CH2CH3 Additions follow Markovnikov's Rule HCl H3C C CCH2CH2CH3 CH COOH 3 H CH2CH2CH3 Cl + H3C Cl H3C CH2CH2CH3 H Additions usually (not always) give trans stereochemistry of H and X HC CCH2CH2CH3 HCl (XS) CH3COOH CH3CCl2CH2CH2CH3 Excess of acid leads to a dihalide product through two consecutive additions. H3C C CCH2CH2CH3 Br2 Br CCl4 H3C CH2CH2CH3 Br Halogens add to alkynes to give addition products with trans stereochemistry... Watch out for further addition when XS reagent is present! Addition of HX and X2 to Alkynes 5 6 Br- Br H H C C H + H+ Br- H C C H H 1st Step: Electrophilic addition of H+ to Alkyne π-bond. H C C H VINYL CATION 2nd Step: Nucleophilic attack of Br- to vinyl cation Markovnikov Addition H R C C R C C H HBr Br Br- H H Br- H C C H C C R H Br H Observed Product C C R H R Alkynes will add the electrophile so as to give the most stable vinyl cation Mechanism of Electrophilic Addition to Alkynes R > R H H R > R ~R H R Most Stable R H H > C C H STABILITY ~ H H H C C H H Least Stable Vinyl Cations are less stable than similarly substituted alkyl carbocations. This accounts for the somewhat lower reactivity of alkynes towards electrophilic reagents (Hammonds Postulate) Vinyl Cation Stability b. Hydration of Alkynes Like the acid-catalyzed addition of water to alkenes, water can be added to alkynes. However, because of the somewhat lower reactivity of the alkyne, the reaction must be carried out with the use of the extremely electrophilic Hg(II) to initially activate the alkyne (see oxymercuration-demercuation of alkenes). The reaction follows a Markovnikov process, with initial production of a vinyl cation which is subsequently trapped by water (acting as a nucleophile) to give an enol (vinyl alcohol). This enol is unstable and undergoes a TAUTOMERIZATION (rearrangement in which a new structural isomer is formed by shift of a hydrogen atom and a π-bond) to form a ketone. Since the reaction follows a Markovnikov regiochemistry, a methyl ketone is produced from the reaction of a terminal alkyne. 7 R C C H R1 C C R2 HgSO4 H+/H2O HgSO4 H+/H2O O R C CH3 O O R1 C CH2R2 + R1H2C C R2 The acid catalyzed hydration of alkynes results in the formation of ketones...... Terminal alkynes methyl ketones Internal alkynes ketone mixture MECHANISM R C C H + Hg+2SO4-2 Hg R C C OH2 H Markovnikov type addition of mercuric ion to alkyne, resulting in formation of most stable vinylic cation -H+ Rapid H O H O Rearrangement C C R C CH3 8 Keq = 10 R H KETO Tautomer H H O Hg C C R H H30+ H O Hg C C R H Tautomers are special kinds ENOL Tautomer of constitutional isomers which can rapidly interconvert. Hydration of Alkynes c. Hydroboration of Alkynes Hydroboration of terminal alkynes is an important reaction for the synthesis of aldehydes. It should be noted that this reaction is complimentary to the mercury catalyzed hydration described just previously. The reagent based specificity allows complete regiochemical (and product) control. R C C H HBR2 THF R1 C C R2 HBR2 THF O R C H O O R1 C CH2R2 + R1H2C C R2 H2O2 NaOH,H2O H2O2 NaOH,H2O The hydroboration of alkynes results in the formation of: Terminal alkynes aldehydes Internal alkynes ketone mixture MECHANISM H BR2 R C C H R C C H + HBR2 HBR2 = disiamylborane A sterically large hydroborating agent; can stop at the mono adduct H2O2 NaOH,H2O H OH R C C H ENOL Tautomer Keq = 106 H O R C C H H KETO Tautomer B H Hydroboration of Alkynes O R C CH3 HgSO4 H3O+ Methyl Ketone R C C H HBR2 THF H2O2 NaOH,H2O H O R C C H Aldehyde H Complimentary Reactions of Terminal Alkynes 5. Catalytic Hydrogenation of Alkynes Alkynes can be catalytically hydrogenated (reduced) to yield alkenes or alkynes. Because the reductions of alkenes and alkynes occur with almost equal facility (actually, alkynes can be reduced slightly easier), it is often difficult to stop the reduction of an alkyne at the alkene stage when using standard catalysts. Complete hydrogenation of the triple bond to yield an alkane is done through the use of two equivalents of H2 and a standard catalyst. Partial reduction of the alkyne (stopping at the alkene stage) can be carried out through the use of a Lindlar Catalyst. In the Lindlar catalyst, a "poison" (usually an aromatic amine such as pyridine or quinoline) has been added to the catalyst to lower its activity, further reduction is prevented. The stereoselectivity of this reaction is determined by SYN addition of the complexed hydrogen to the sme face of the alkyne π-bond. Cis alkenes are formed. 8 9 H2, Pd/C EtOH R C C H RH2C CH2 + R CH2 CH3 + UNREACTED ALKYNE The catalytic reduction of alkynes, using standard hydrogenation catalysts gives a mixture of products: alkenes (from one equivalent of H2 uptake), alkanes (from two equivalents of H2 uptake) and unreacted alkyne (all H2 used up). The use of two equivalents of H2 will give only alkanes 2 H2, Pd/C EtOH R C C H R CH2 CH3 Catalytic hydrogenation of alkynes can be stoped at the alkene stage through the use of Lindlar Catalysts (Deactivated Catalyst) H2 Lindlar catalyst R C C H Lindlar catalyst : Pd/Caso4 deactivated by addition of poison (quinoline) RH2C CH2 Catalytic Hydrogenation of Alkynes R1 C C R2 H2 Lndlar catalyst R1 R2 C C H H SYN addition leads to cis stereochemistry R1 H2 Metal surface attraction weakens H-H bond C H C R2 H Activated H2 adds to alkyne π-system R1 R2 C C H H + Catalyst regenerated SYN Addition of Alkynes 6. Dissolving Metal Reductions of Alkynes In contrast to the catalytic hydrogenation method, which yields cis-alkenes from alkynes, the reaction of alkynes with sodium or lithium metal in liquid ammonia produces the complimentary trans-alkene. The mechanism of this reaction involves the addition of electrons (given up by the electropositive metal) to the π-system of the alkyne to from an intermediate radical-anion which reacts with the ammonia solvent by pulling off a proton. The resulting radical, in turn, undergoes further reduction to form a vinyl anion. Which again reacts with ammonia, by pulling off a proton, to give the final trans-alkene. The trans nature of the reaction occurs because in the radical anion intermediate, the orbital 10 with the lone-pair and the orbital with the single electron want to be as far apart as possible (in order to minimize repulsions). Thus they take on a trans arrangement. R1 H 1) Li, NH3 C C R2 2) H2O R2 R1 Trans only H "Dissolving Metal" Li + NH3 Li+ + e- (NH3)n Electropositive metals such as Li, Na will ionize in a liquid ammonia solution, giving up an electron which becomes "solvated " by ammonia molecules Mechanism R1 solvated R1 C C R2 electrons NH3 R2 R1 as an acid R1 H H R2 NH2 + + NH2- R2 Radical Anion The radical and lone-pair get as far apart as possible - H solvated electrons reduction of radical to anion NH3 as an acid R1 H R2 Dissolving Metal Reduction of Alkynes to trans Alkenes 7. Oxidative Cleavage of Alkynes Powerful oxidizing agents such as ozone or acidic KMnO4 will cleave the triple bonds of alkynes. The triple bond is generally less reactive than is the double bond so yields of this reaction may be low. If a terminal alkyne is utilized in this reaction, a carboxylic acid and CO2 is produced. R C C H R1 O3 or KMnO4 O3 or C C R2 KMnO 4 O R C OH + CO2 R1 O C OH + R2 O C OH Alkynes undergo oxidativecleavage just like alkenes... However, they are somewhat less reactive and the yields of cleavage products can be low. Oxidative Cleavage of Alkynes 8. Alkylation of Alkynes a. Acidity of Alkynes One of the striking differences in the chemical behavior of alkenes and alkynes is their differences in acidities. Terminal alkynes have hydrogens which are many times more acidic than the hydrogens of alkanes or alkenes. This is a result of the increased s character in the sp bond between the carbon and hydrogen of terminal alkynes. The anion ( which is the conjugate base of a terminal 11 alkyne) is more stable because the electron pair is closer to the positive charged carbon nucleus. Thus, the acidic hydrogen of terminal alkynes can be removed by a strong base (such as NH2-) to form the acetylide anion : R-C≡C:Hydrocarbon Ka pKa HC C H 10-25 25 H2C CH H 10-44 44 H3CH2C H 10-60 60 Conjugate Base Stronger Acid HC C More Stable H2C CH Weaker Acid H3C H2C Less Stable Carbon centers with increasing s orbital character will best stabilize carbanions because the negative anion is closer to the positive nucleus. Acetylide Formation: A strong base is able to remove a terminal hydrogen from an alkyne resulting in formation of the conjugate base called acetylide anion. R C C H NaNH2 NH3 R C C Na+ + NH3 NH2- = Amide Anion... A very strong base (about 1015 x stronger than HO-) NH3 pKa = 35 Acidity of Alkynes b. Alkylation of Acetylides By using the parent acetylene or a terminal alkyne, it is possible to introduce one, or two alkyl groups and thus form an entirely new alkyne. The presence of an unshaired pair of electrons on an acetylide anion makes the carbon strongly nucleophilic. Thus acetylide anions ( R-C≡C:-) can react with alkyl halides such a bromomethane (CH3-Br) to substitute for the halogen a yield a new alkyne product ( R-C≡C-CH3). H C C H KNH2 NH3 Amide anion abstracts proton...forms the acetylide anion. H3C Br H C C Acetylide acts as a nucleophile...forms a new carbon-carbon bond (Alkylation) A second alkylation step leads to a new H C C C CH 3 carbon carbon bond. 3 Alkylation of Alkynes ∂+ ∂- KNH2 NH3 Amide again abstracts a proton Br- CH +3 ∂ H C C CH3 + Br- ∂ C C CH3 12 O CH3CCH3 Cl PCl5 CH3CCH3 NaNH 2 Cl ∆ Br Br CHCCH3 Br Br CO2 HgSO4 H2 O H2 SO4 (1) O3 (2) Zn / H2 O HC CCH3 NaNH 2 liq NH3 2 HCl 2 Br2 O CCH3 HO + 1-propyne Cu(NH 3 )4 OH + - Cu C CCH3 + - Na CH3 CH2 Br Ag(NH3 )2 OH NaNH 2 ∆ CH3CH2C CCH3 + - Br Br CH2CHCH3 Ag C CCH3 CH3 Li in liq NH3 CH3C CCH3 H C C H CH3 H H H2 , Ni 2 B C C or CH CH3 3 Pd/CaCO3 H2 , Quinoline Alkyne Reaction Summary C CCH3