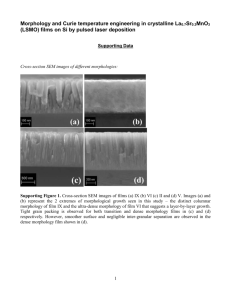
Fabrication of Structured Polymer Films Using Vapor Deposition
advertisement

i Fabrication of Structured Polymer Films Using Vapor Deposition Techniques by Xichong Chen Submitted in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Supervised by Professor Mitchell Anthamatten Department of Mechanical Engineering Materials Science Program Arts, Sciences and Engineering Hajim School of Engineering and Applied Sciences University of Rochester Rochester, New York 2009 ii CURRICULUM VITAE The author was born in Urumqi, China on August 4, 1977. He attended Fudan University from 1995 to 1999 and matriculated with a Bachelor of Science degree in 1999. He then continued his education at Fudan University and graduated with a Master of Science degree in 2002. He came to the University of Rochester in the Fall of 2003 and began graduate studies in the Materials Science Program at Department of Chemical Engineering. He received a Master of Science degree in Materials Science in 2005. He then pursued his doctoral research in structured vapor-deposited polymer films under the direction of Professor Mitchell Anthamatten. iii ACKNOWLEDGEMENTS It is difficult to overstate my gratitude to my Ph.D. research advisor, Professor Mitchell Anthamatten for his support and guidance of my studies. With his enthusiasm, his inspiration, and his great efforts to explain things clearly and simply, he helped to make research fun for me. Throughout my thesis-writing period, he provided encouragement, sound advice, good teaching and lots of good ideas. I would have been lost without him. I am grateful to the other members of the Anthamatten group. I thank Jiahui Li, Supacharee Roddecha, Alex Papastrat, Kavya Ramachandra, Shuai Zhu for providing a simulating and fun environment in which to learn and grow. I would like to thank my officemate Michelle Wrue for taking charge of lab safety and making my lab experience more enjoyable. I also thank my lab partner Zachary Green, a former member of the Anthamatten group, for his encouragement and accompany in the past two years. I would like to thank Mark Bonino for setup of PBO deposition system, Christine Pratt of the Department of Mechanical Engineering for assistance with nano-indentation and WAXS, Debra Blondell at Eastman Kodak for the vacuum TGA experiments, Julie Smyder of Department of Chemistry and Huanjun Ding of Department of Physics and Astronomy for the AFM measurements. I also appreciate the financial support in the form of a Frank J. Horton Research Fellowship from the Laboratory of Laser Energetics. iv Lastly, I would like to thank my family. The encouragement and support from my beloved wife Lijun Zou and our son, Eric is a powerful source of inspiration and energy. And most importantly, I wish to thank my parents, Suoxian Zhang and Jinfeng Chen. They raised me, supported me, taught me, and loved me. To them I dedicate this thesis. v ABSTRACT New techniques to fabricate structured polymer films using chemical vapor deposition were developed and studied. Two different vapor deposition approaches, one using step-growth polymerization and another using chain-growth polymerization, were employed. The primary objective of this thesis was to determine the feasibility of controlling morphology of vapor-deposited polymer films by introducing non-reactive, immiscible components into the vapor deposition process. Poly(amic acid)s, condensation polymers and precursors to rigid-rod polybenzoxazoles (PBO), formed upon co-deposition of 3, 3’-dihydroxybenzidine (DHB) and pyromellitic dianhydride (PMDA). Deposited coatings were cured under inert gas conditions and resulted in the conversion to semi-aromatic polybenzoxazoles at around 550 °C. Physical and chemical changes occurring during the curing process were studied with FT-IR, TGA and nanoindentation experiments. Successful fabrication of PBO films provided a platform to study simultaneous film growth and phase separation in vapor-deposited condensation polymers. Control of morphology of vapor-deposited condensation polymer films was achieved by the fabrication of polyimide/CuPc composite films, which were made from the co-evaporation of 4,4’-oxydianiline (ODA) and 3,3’,4,4’-biphenyl tetracarboxylic dianhydride (BPDA) in the presence of non-reactive, third-component CuPc. Spectroscopy experiments confirm the formation of polyimide segments and suggest that embedded CuPc molecules have less mobility compared to pure CuPc films. Electron microscopy and XRD studies show evidence of embedded CuPc vi particles at the surface and in the bulk of fine lateral structure with a length scale of about 100 nm. Fabrication of poly (methyl methacrylate) (PMMA) films using initiated chemical vapor deposition (iCVD) provided a platform to investigate film growth and phase separation for a classical chain-growth polymer. An axisymmetric, multi-component iCVD apparatus was designed to study the vapor-phase growth of glassy PMMA films. Key reactor operating parameters, including the hot-zone temperature, reactor base-pressure, substrate temperature, and the monomer/initiator molar feed ratio were systematically varied to understand film growth kinetics. The non-reactive solvent-vapor, t-butanol, was then introduced into the deposition process to promote polymer film dewetting. When solvent-vapor is used, non-equilibrium dewetted structures comprising of randomly distributed polymer droplets were observed. The length-scale of observed topographies, determined using power spectral density (PSD) analysis, ranges from 5 to 100 microns and can be influenced by deposition conditions, especially the carrier gas and solvent-vapor flowrates. Control over lateral length-scale is demonstrated by preparation of hierarchal “bump-on-bump” topographies. Autophobic dewetting of PMMA from SiOx/Si substrate during iCVD process is attributed to a thin film instability driven by both long range van der Waals forces and short range polar interactions. vii TABLE OF CONTENTS Chapter 1. 1.1 1.2 1.3 1.4 1.5 Introduction and Background ..............................................................1 Chemical vapor deposition (CVD) .....................................................1 Condensation chemical vapor deposition ...........................................4 Initiated chemical vapor deposition (iCVD).......................................7 Film morphology ..............................................................................11 Objective and outline ........................................................................17 Chapter 2. 2.1 2.1.1 2.2 2.2.1 2.2.2 2.2.3 2.2.4 2.2.5 2.3 2.3.1 2.3.2 2.3.3 2.3.4 2.4 Vapor Deposition and Curing of Polybenzoxazole Precursors .......20 Introduction.......................................................................................20 Polybenzoxazoles (PBOs).................................................................20 Experimental .....................................................................................24 Materials ...........................................................................................24 Vapor deposition polymerization of PBO precursor 3 .....................24 Solution synthesis of PBO precursor 3 .............................................25 Curing of PBO precursors.................................................................26 Characterization ................................................................................26 Results and discussion ......................................................................27 Sublimation of PMDA and DHB monomers ....................................27 Change of chemical structure upon curing .......................................29 Thermal properties ............................................................................33 Mechanical properties of PBO..........................................................34 Summary ...........................................................................................39 Chapter 3. Morphology of Vapor Deposited Polyimides Containing Copper Phthalocyanine .....................................................................................41 Introduction.......................................................................................41 Polyimides.........................................................................................41 Copper phthalocyanine .....................................................................42 Organic photovoltaics (OPV) ...........................................................44 Experimental .....................................................................................46 Materials ...........................................................................................46 Substrate preparation ........................................................................47 Vapor deposition of polyimide/CuPc composites.............................47 Characterization ................................................................................49 Results and discussion ......................................................................50 VDP of polyimides ...........................................................................50 VDP of CuPc/polyimide hybrid films...............................................56 Morphology of composite films........................................................60 Summary ...........................................................................................68 3.1 3.1.1 3.1.2 3.1.3 3.2 3.2.1 3.2.2 3.2.3 3.2.4 3.3 3.3.1 3.3.2 3.3.3 3.4 viii Chapter 4. 4.1 4.1.1 4.1.2 4.2 4.2.1 4.2.2 4.2.3 4.3 4.3.1 4.3.2 4.3.3 4.3.4 4.3.5 4.3.6 4.4 Chapter 5. 5.1 5.1.1 5.2 5.2.1 5.2.2 5.2.3 5.3 5.3.1 5.3.2 5.4 Multi-component Vapor Deposition Polymerization of Poly(Methyl Methacrylate) in an Axisymmetric Vacuum Reactor ..................................................................................................70 Introduction.......................................................................................70 Materials and application..................................................................70 iCVD reactor design .........................................................................71 Experimental .....................................................................................77 Materials ...........................................................................................77 Deposition of PMMA films ..............................................................78 Characterization of iCVD PMMA films...........................................80 Results and discussion ......................................................................81 iCVD of PMMA films using base-case conditions...........................81 Hot zone configuration and temperature...........................................86 Reactor base pressure........................................................................90 Substrate temperature........................................................................94 Monomer/initiator molar feed ratio ..................................................95 Reactor modifications .......................................................................97 Summary ...........................................................................................99 Solvent-Assisted Dewetting During Chemical Vapor Deposition Process.................................................................................................101 Introduction.....................................................................................101 Dewetting and solvent-induced dewetting......................................101 Experimental ...................................................................................106 Materials .........................................................................................106 Initiated chemical vapor deposition of PMMA ..............................107 Characterization of iCVD films ......................................................107 Results and discussion ....................................................................109 Dependence of film morphology on deposition conditions............109 Proposed Dewetting Mechanism ....................................................122 Summary .........................................................................................130 Chapter 6. Summary and Future Work .............................................................132 6.1 Summary .........................................................................................132 6.2 Future work.....................................................................................135 References................................................................................................................138 ix LIST OF TABLES Table 2.1 Molar loss ratio at different operating conditions..................................29 Table 2.2 FT-IR peak assignments for vapor-deposited poly (amic acid), polyimide and PBO. ..............................................................................31 Table 3.1 Summary of typical VDP attempts to grow polyimide films. ...............52 Table 3.2 FT-IR peak assignments for CuPc and vapor-deposited BPDA-ODA polyimide...............................................................................................56 Table 4.1 Deposition conditions of iCVD experiments.........................................79 Table 5.1 Parameters from fitting k-correlation model for PSD plot of dewetted and stable films....................................................................................119 x LIST OF FIGURES Figure 1.1 Close-up of chemical vapor deposition (CVD) reactor..........................2 Figure 1.2 Schematic drawing of VDP processes with different polymerization mechanisms. a) Step polymerization; b) chain polymerization; c) activated monomer polymerization; d) activated substrate polymerization. ......................................................................................3 Figure 1.3 General two-step imidization reaction....................................................5 Figure 1.4 Schematic drawing of initiated chemical vapor deposition (iCVD) technique. ...............................................................................................8 Figure 1.5 Schematic drawing of iCVD processes using different dissociate methods. .................................................................................................9 Figure 1.6 Scheme for free-radical polymerization in iCVD process. ..................10 Figure 1.7 Diagram of growth effects including diffusion, shadowing and reemission which may affect surface morphology during thin film deposition.............................................................................................12 Figure 1.8 Principle of polymerization-induced phase separation.........................16 Figure 2.1 Structural comparison of fully-aromatic (1) and partially-aromatic PBO(2). ................................................................................................21 Figure 2.2 Synthesis of semi-aromatic PBO involving thermal rearrangement of polyimide precursor. ............................................................................23 Figure 2.3 Schematic diagram of the deposition system inside the vacuum chamber................................................................................................25 Figure 2.4 Vapor pressure curves of PMDA and DHB from vacuum TGA measurement. .......................................................................................28 Figure 2.5 FT-IR Spectra of (a) as-deposited film (b) film cured at 200 °C for 5 hours, and (c) film cured at 550 °C for 1 hour. The spectrum in (c) was taken using reflective mode. ................................................................32 Figure 2.6 TGA thermogram of as-deposited poly (amic acid) film. ....................34 Figure 2.7 (a) Modulus and (b) hardness of vapor-deposited and solution-cast films at different stages of curing corresponding to poly(amic acid) (PAA), polyimide (PI), and polybenzoxazole (PBO). .........................36 Figure 2.8 Effect of curing rate on the mechanical properties of cured polyimide: (a) modulus and (b) hardness...............................................................38 Figure 3.1 Molecular structure of copper phthalocyanine (CuPc).........................43 Figure 3.2 Two types of OPV devices: (a) Schottky-type junction and (b) pn-heterojunction. ................................................................................45 Figure 3.3 Three-component VDP reactor for depositing polyimide/CuPc composite. ............................................................................................48 xi Figure 3.4 Vapor pressure curves of BPDA, ODA and PMDA, obtained by vacuum TGA measurement. ................................................................51 Figure 3.5 Reaction of BPDA and ODA to form poly(amic acid) and subsequent curing to yield polyimide. ....................................................................54 Figure 3.6 FT-IR extinction coefficients of a) cured BPDA-ODA polyimide; b) heat-treated CuPc; and c) a composite polyimide/CuPc film. .............55 Figure 3.7 Optical absorption spectra of: a) pure BPDA-ODA polyimide and CuPc films; b) Q-band of CuPc and composite polyimide/CuPc films. .......58 Figure 3.8 TEM images of vapor deposited BPDA-ODA poly(amic acid) and cured polyimide films containing about 30 and 60 vol % CuPc..........61 Figure 3.9 Atomic force micrographs of a) BPDA-ODA polyimide, b) 30 % CuPc in polyimide, c) 60 % CuPc in polyimide, and d) pure CuPc films. Film thicknesses for a-d) were measured to be 400, 264, 80, and 141 nm, respectively. ..................................................................................65 Figure 3.10 Temperature dependent X-ray diffraction spectra of vapor-deposited films: a) pure CuPc, and b) 60 vol % CuPc in BPDA-ODA polyimide. ............................................................................................................67 Figure 4.1 Molecular structure of poly (methyl methacrylate) (PMMA)..............70 Figure 4.2 A schematic drawing of the pancake-shape iCVD reactor setup for coating on flat substrates. This figure was reproduced from reference 124........................................................................................................73 Figure 4.3 Schematic drawing of axisymmetric iCVD setup. ...............................74 Figure 4.4 (a) Top-view of 11-inch aluminum base-plate; (b) schematic of custom-built, axisymmetric iCVD apparatus.......................................76 Figure 4.5 Hot-zone configurations employed in iCVD experiments. ..................80 Figure 4.6 Thickness profile of as-deposited PMMA film (8-hour deposition) on a 3-inch-diameter silicon wafer (unit: nanometer). ................................83 Figure 4.7 SEM image of as-deposited PMMA film.............................................83 Figure 4.8 FT-IR spectrum of PMMA deposited using axisymmetric iCVD reactor. .................................................................................................84 Figure 4.9 Three steps in iCVD of PMMA process. 1) Thermal initiation; 2) free radical and monomer transport; 3) condensation and reaction. ...........86 Figure 4.10 PMMA deposition rates for different reactor hot-zones in axisymmetric iCVD reactor operating under base-case conditions. ..87 Figure 4.11 Logarithmic plot of PMMA deposition rate versus hot-zone temperature.........................................................................................89 Figure 4.12 Experimental results (○) and model fit (dashed line) illustrating the relationship between deposition rate and reactor base pressure.........90 Figure 4.13 Simplified plug-flow reactor diagram involving one reactor for thermal initiation and a second reactor for recombination.................91 Figure 4.14 Plot showing deposition rate dependence on substrate temperature. .94 Figure 4.15 Size exclusion chromatograms of vapor deposited PMMA: a) effect of monomer/initiator molar feed ratio, and b) effect of non-reactive third component, t-butanol..........................................................................96 xii Figure 4.16 A hot-zone nozzle was added to improve deposition rate. a) Original hot-zone design; b) reactor modification with hot-zone nozzle. ........98 Figure 4.17 The dependence of deposition rate on solvent flow rate in base-case condition.............................................................................................99 Figure 5.1 Schematic drawing of a modulated surface for liquid film on solid substrate. ............................................................................................103 Figure 5.2 Different stages for a typical dewetting process.The length of the bar is 100 µm. This figure was reproduced from reference 133..................104 Figure 5.3 Interferometry images of as-deposited PMMA films: (a) deposition without solvent; (b) deposition with t-butanol solvent (S10-N5); and (c) the same film, following annealing at 120 °C for 2 hours. ..........110 Figure 5.4 Dependence of average mass deposition rate on solvent and carrier gas flow rate. The introduction of carrier gas independently increases deposition rate. Solvent-vapor flow increases deposition rate, and lowers viscosity at the surface of the growing film. ..........................113 Figure 5.5 As-deposited film morphology at different solvent-vapor and carrier gas flowrates. .....................................................................................115 Figure 5.6 Power spectra density of films deposited with: a) different carrier gas (N2) and b) different solvent-vapor flowrates. Dashed lines are least-squares fits to the k-correlation model. .....................................118 Figure 5.7 Process diagram indicating whether dewetting was observed in solvent-vapor assisted iCVD growth of glassy PMMA films. Depositions were performed on a) plasma-treated silicon wafer substrates and b) hydrophobically modified substrates. The symbols × and ○ indicate dewetted and wetted films respectively. ..................120 Figure 5.8 Proposed dewetting mechanism. Initially a liquid layer (yellow) adsorbs on the surface consisting of predominately methyl methacrylate and t-butanol. As PMMA is polymerized, a strongly anchored layer of PMMA (blue) coats the substrate. The solvent-monomer mixture forms an unstable film on top of the anchored PMMA. (a) If deposition is slow, or if the viscosity is low enough, then surface fluctuations reach the substrate and dewetting occurs, leaving glassy PMMA droplet-like features (green). (b) If deposition is fast enough or the viscosity is high enough, then the time-scale of dewetting is too slow and a uniform coating, which may possess small surface undulations, results. ........................................123 Figure 5.9 Topography (left) and phase (right) images for (A, a) dewetted PMMA film on Si/SiOx substrate; (B, b) dip-coated (and partially dewetted) PMMA on bare silicon wafer.............................................................125 Figure 5.10 Topography of a “bump-on-bump” structure measured using interferometry. ..................................................................................130 xiii LIST OF SYMBOLS AND ABBREVIATIONS SYMBOLS Poisson’s ratio E Young’s modulus parallel to the surface E Young’s modulus perpendicular to the surface λ Wavelength θ Bragg diffraction angle Number average molecular weight Mn Mw Weight average molecular weight ki Initiation rate constant Ci Gas concentration of initiator LHZ Hot-zone length Initial inlet initiator concentration Ci0 v0,HZ Total volumetric flow rate of the hot-zone ax Reactor cross-sectional area Xi Degree of initiator conversion kr Recombination rate constant Concentration of free radical species Ci LRZ Length of the recombination zone Degree of free radical recombination Xr Tsub Substrate temperature Thot Hot-zone temperature Re Reynolds number ρ Gas density Gas velocity vz D Diameter of reactor µ Dynamic viscosity Aeff Effective Hamaker constant S The spreading coefficient h Film thickness u Amplitude of undulation q Wavevector R Fluctuation growth rate Liquid viscosity Molecular length scale a2 Surface tension Pi Gas-phase pressure xiv ABBREVIATIONS CVD VDP PMDA ODA UHV iCVD HF-CVD PE-CVD piCVD gCVD AFM TEM SEM PIPS PMMA PBO CuPc TBPO PPA DHB FT-IR TGA DMAc KBr PAA PI MLR 6FDA NTDA DMDA OPV OLED BPDA XDA PTDA UV/Vis QCM RMS WAXS TIPS CFD CW Chemical vapor deposition Vapor deposition polymerization Pryomellitic dianhydride 4,4’-Oxydianiline Ultra high vacuum Initiated chemical vapor deposition Hot-filament chemical vapor deposition Plasma-enhanced chemical vapor deposition Photoinitiated chemical vapor deposition Grafting chemical vapor deposition Atomic force microscopy Transmission electron microscopy Scanning electron microscopy Polymerization-induced phase separation Poly(methyl methacrylate) Polybenzoxazole Copper phthalocyanine tert-Butyl peroxide Poly(phosphoric acid) 3,3’-Dihydroxy-benzidine Fourier transform infrared spectroscopy Thermogravimetry analysis N, N-Dimethylacetamide Potassium bromide Poly(amic acid) Polyimide Molar-loss ratio 2,2-bis(3,4-dicarboxyphenyl) hexafluoropropane dianhydride Naphthalene tetracarboxylic dianhydride Decamethylene diamine Organic photovoltaics Organic light-emitting diodes Biphenyl tetracarboxylic dianhydride Xylene diamine Perylene tetracarboxylic dianhydride Ultraviolet/visible Quartz crystal monitor Average root-mean-square Wide-angle X-ray scattering Thermal-induced phase separation Computational fluid dynamics Cooling water xv NMP HPLC GPC THF PDI TEA HZ RZ MMA PSD N-Methylpyrrolidone High pressure liquid chromatography Gel permeation chromatography Tetrahydrofuran Polydispersity index Triethylamine Hot-zone Recombination zone Methyl Methacrylate Power spectral density 1 Chapter 1. Introduction and Background 1.1 Chemical vapor deposition (CVD) Chemical vapor deposition (CVD) is a versatile chemical process used in the production of coatings1, powders2, fibers3 and monolithic components4. This method is usually employed to form dense structural parts or coatings using the reaction or decomposition of gases with relatively high vapor pressure. In a typical CVD process, the substrate is exposed to one or more volatile precursors usually in the gas phase, which react and/or decompose on the substrate to produce the desired deposit. CVD is a complicated process and Figure 1.1 shows a close-up of a typical CVD reactor. Mass transport occurs first after precursor gas is introduced into the reactor. The mass transport could be gas flow which follows streamline through the entire reactor, or the mass diffusion driven by the concentration gradient. Although CVD is generally conducted in a low pressure, vacuum environment, gas phase chemistry may still happen between precursor molecules or electrons and ions from plasma. The most important part in a CVD process is surface chemistry which includes the formation of the product films, removal of byproducts after reaction and ion bombardment if plasma is involved. Vapor processing technique is well suited to modify surface properties of bulk materials, for example scratch-resistant and antimicrobial coatings1, or to prepare thin film devices such as thin film transistors5, 6, photovoltaic cells7, 8, and gas separation membranes9. 2 Figure 1.1 Close-up of chemical vapor deposition (CVD) reactor. A key advantage of the CVD process lies in the fact that the reactants used are gases. Therefore, vapor phase processing is differentiated from solution-based techniques in many ways. These include: 1) film growth is generally slow (~ µm/hr), enabling films to be grown to precisely desired thicknesses; 2) film properties can be tuned by adjusting the molar flux ratios of gaseous precursor components; 3) polymers can be coated onto rough substrates or even onto complex 3D geometries such as spheres10, 11 ; and 4) vapor-derived processes are “solventless” and are attractive from an environmental standpoint.12 CVD processes have also been widely used to prepare functional polymer coatings. Vapor deposition polymerization (VDP) of both condensation and addition polymers have been performed in the absence of plasma and resulted in polymer chains that are chemically similar to their solution analogs. Generally, VDP can be 3 briefly summarized into four categories depending on the polymerization reaction mechanism.13 They are (1) step polymerization; (2) chain polymerization; (3) activated monomer polymerization; and (4) activated substrate polymerization. In step-growth VDP, two or more monomers with reactive groups are vaporized and transport to target substrate. Then condensation polymerization between these monomers occurs to form polymer film (Figure 1.2 a). In chain-growth VDP, an initiator is used and dissociated in the gas phase. The activated species and monomer transport to the substrate where chain growth polymerization happens (Figure 1.2 b). For the activated monomer polymerization, monomer itself is activated by thermal or electromagnetic energy to generate activated species. These species undergo chain linking or crosslinking reaction to form polymer (Figure 1.2 c). Alternatively, the target substrate can be activated before the exposure of monomer vapor. Then the reaction between activated substrate and reactive monomer results in single monolayer of polymer film (Figure 1.2 d). a) c) b) MA MB M d) M I I I * M M M * *********** Figure 1.2 Schematic drawing of VDP processes with different polymerization mechanisms. a) Step polymerization; b) chain polymerization; c) activated monomer polymerization; d) activated substrate polymerization. 4 In this study, condensation chemical vapor deposition using step-growth mechanism and initiated chemical vapor deposition using chain-growth mechanism are used to deposit polymer films and will be introduced in detail in the next two sections, respectively. 1.2 Condensation chemical vapor deposition Condensation chemical vapor deposition usually involved vaporizing two different polyfunctional (a polyfunctional monomer is one with two or more functional groups per molecules) monomers in which each monomer possesses only one type of functional group. The monomer molecules in the gas phase transport to the target substrate, then the step-growth polymerization reaction on the substrate can be represented in a general manner by the equation: n A A nB B ( A A B B) n [1.1] where A and B are the two different types of functional groups. For a typical vapor deposition system, it consists of a vacuum chamber equipped with two separately heatable monomer sources, a system to control the fluxes from the sources, a shutter system and a sample holder. Depending on the operating system pressure, two different types of source cells have been used. Resistively or radiatively heated quartz boats or tubes are often used under a system pressure of P ≥ 10-4 Pa (7.5×10-7 Torr) and are designed for high deposition rates. For an ultra-high-vacuum (UHV) environment, Knudsen cells are used to allow low 5 deposition rates and to minimize the contamination of the UHV system with organic materials.14 Condensation polymers including polyamides15, polyimides16-19, and particularly in our group’s laboratory, polybenzoxazoles20, have been successfully fabricated using VDP techniques. However, the majority of these studies have been focused on polyimides. Aromatic polyimide thin films were first vapor-deposited independently by Iijima et al 17 and Salem et al 18 in 1985 and 1986, respectively. It involves the co-evaporation of two reactive monomers, Pryomellitic dianhydride (PMDA) and 4,4’-oxydianiline (ODA) onto a substrate under vacuum conditions, followed by a curing step ( >200°C ) (Figure 1.3). O O O O + O O H2N NH2 O RT O O HO O NH H N OH O O -2nH2O O O N N O n 200°C O O n Figure 1.3 General two-step imidization reaction. 6 Condensation CVD process is usually performed at pressures below 10-6 Torr. Due to the large mean free path (>10 m) at this pressure, the gas molecules can be transported from source to substrate without hitting each other which preclude gas-phase reaction between feed components. The flux from each monomer source has to be balanced to fabricate high quality film. This can be accomplished by thermal or rate control of evaporate sources. In addition, the monomer involved in the condensation CVD process requires high reactivity, higher than solution or melt step-growth polymerization reactions. Compared with solution-casting films, polymer films from condensation CVD process can have different properties which strongly depend on processing conditions. For example, VDP polyimides usually have higher density, stiffness but less hydroscopic then solution-cast polyimides.13 Polymer films from condensation vapor deposition are usually glassy, nearly isotropic with less orientation than is typically observed in solution processed films.21 One of the limitations for condensation CVD is the resulting macromolecules are lower in molecular weight than their solution analogs.22 VDP involves a solid-state polymerization that, in the absence of solvent, is kinetically hindered due to low monomer and oligomer mobility.16 In order to deposit relatively high molecular weight polymer, different approach, i.e. initiated chemical vapor deposition can be used by introducing highly reactive free radical species. This technique will be discussed in the next section. 7 1.3 Initiated chemical vapor deposition (iCVD) Unlike condensation chemical vapor deposition, initiated chemical vapor deposition (iCVD) is a relatively new deposition technique to make polymer thin films using chain-growth mechanism.23 iCVD method could be regarded as a subset of a common technique called hot filament chemical vapor deposition (HF-CVD). The central part of the HF-CVD setup is a metal filament (typical tantalum or tungsten). Thermal decomposition is achieved by flowing the gas species past resistively-heated filament wires, and the activated species generated by this process undergo polymerization reactions to form a film on the cooled substrate. The deposition is usually performed at reduced pressure and the substrate is placed within a small distance (2~3 cm) from the filament. Because there are fewer reaction pathways in HF-CVD than in the plasma-enhanced CVD (PE-CVD) process, HF-CVD can provide more well defined chemical structures of the as-deposited films and minimize generation of structural damage in the films.24, 25 iCVD technique was developed by Gleason et al of Massachusetts Institute of Technology in late 1990’s.25 It differs from conventional HF-CVD on one main aspect: an initiator in addition to the monomer is introduced into the vacuum chamber. Mao and Gleason have demonstrated that iCVD can produce a variety of functional polymer such as poly (tetrafluoroethylene)26, poly (glycidyl methacrylate)27, poly (2-hydroxyethyl methacrylate)28, poly (methyl methacrylate)29 and poly (perfluoroalkylethyl methacrylate)30. In a typical iCVD process, monomer species and initiator are vaporized and fed at moderately low pressure (~0.1-10 Torr) 8 over array of resistively-heated Nichrome filament (~250-400°C) and across a cooled substrate. Monomer molecules absorb onto the hot filament and then reemit without reaction. Initiator molecules undergo hemolytic bond cleavage and desorb as free radical species. Both monomer and initiators diffuse and adhere to the substrate. Then solid state free-radical polymerization occurs to form linear chains (Figure 1.4). Figure 1.4 Schematic drawing of initiated chemical vapor deposition (iCVD) technique. Several CVD techniques have been developed based on the principles of iCVD, as illustrated in Figure 1.5. Photoinitiated chemical vapor deposition (piCVD) uses a volatile photoinitiator to initiate free-radical polymerization of gaseous monomers under UV irradiation.31 This technique simplifies the reactor design and eliminates any heat radiation from hot filament. Grafting Chemical vapor deposition (gCVD) uses type II photoinitiator to graft polymer chains to the substrate surface.32 9 Resulting grafted polymer layer is resistant to abrasion and is stable against virtually any solvent. Our laboratory uses a packed hot-zone as an alternative to the hot filament to dissociate free radical initiator.33 This design enables poly(methyl methacrylate) (PMMA) to be deposited at relatively low dissociate temperature (~200 °C) compared with hot filament design (>450 °C)29. Figure 1.5 Schematic drawing of iCVD processes using different dissociate methods. The mechanism of iCVD polymerization is believed to occur through three major steps, (1) decomposition of an initiator in the vapor phase to form primary free radicals, (2) diffusion and adsorption of primary radicals and monomer from the vapor phase onto a cooled surface, and (3) polymerization of monomer on the surface to form a continuous polymer coating. The primary radicals first initiate chain polymerization on the substrate surface by adding to first one monomer unit (initiation), then these active radical centers propagate by adding more and more 10 monomer units to form longer polymer chain (propagation). These polymer chains could terminate by coupling or disproportionating with other polymer chain or reactive radical species (termination) 34 (Figure 1.6). Because of the presence of free-radical initiator, deposition rate of the iCVD process can excess 10 µm/hr. Due to the high reactivity of free radical species, resulting polymers have molecular weights range from 15 K to 50 K Daltons, which could be hardly achieved in the condensation VDP process. Radical Generation (filament/UV) I 2 2I kd Initiation I M IM ki Propagation IM n M IM n 1 kp Termination IM n IM p IM n p I kt IM n IM p IM n IM p kt Figure 1.6 Scheme for free-radical polymerization in iCVD process. The reaction kinetics of iCVD process has been investigated and it is found that it can be succinctly considered as a free radical polymerization in the bulk yet without the presence of a liquid phase.35 However, it is distinct in that, unlike bulk thermal polymerization, iCVD effectively separates the initiator decomposition from the polymerization events (propagation and termination). This additional degree of freedom should, in principle, allow a greater degree of control over processing and 11 resulting coating properties. It also provides greater processing flexibility because a lower substrate temperature could be employed to coat heat-sensitive materials such as plastics, fabrics, and pharmaceutics. 1.4 Film morphology Thin film deposition including chemical vapor deposition is the most ubiquitous and critical part of the processes used to manufacture high-tech devices such as microprocessors36, memories37, solar cell38, microelectromechanical systems (MEMS)39, lasers40, solid-state lighting41, and photovoltaics42. The morphology and microstructure of these thin films directly controls their optical43, magnetic44, and electrical properties44, which are often significantly different from bulk material properties. Precise control of morphology and microstructure during thin film growth is paramount to producing the desired film quality for specific applications. To date, many thin film deposition techniques have been employed for manufacturing films, including thermal evaporation45, sputtering deposition46 and chemical vapor deposition47. Many deposition factors contribute to the formation of complex morphology on the surface of a film. First, during deposition process, there is always random noise that exists naturally because atoms do not arrive at the surface uniformly. Noise competes with surface smoothing process, such as surface diffusion, to form a rough morphology if the deposition is performed at a high growth rate. In addition, growth 12 roughness can also be increased by some growth processes such as geometrical shadowing.48 Shadowing is a result of deposition by non-normal incident flux.49 In both sputtering deposition and chemical vapor deposition, atoms do not always approach the surface in parallel and they arrive at the surface with a distribution of trajectories very often. Another important effect to consider is the sticking coefficient which is defined as the probability that a particle will stick to the surface when it strikes.50 The sticking coefficient may not be equal to unity which means particle is allowed to reemit from the surface upon impact. The particle may then deposit on the different location which leads to a smoothing effect. Common growth effects which contribute to the overall film morphology are summarized in Figure 1.7. Figure 1.7 Diagram of growth effects including diffusion, shadowing and reemission which may affect surface morphology during thin film deposition. There are two classes of measurement techniques which allow for a collection of quantitative information about film morphology: real-space imaging techniques, 13 and diffraction techniques.51 Real-Space imaging techniques include atomic force microscopy (AFM), transmission electron microscopy (TEM), scanning electron microscopy (SEM), stylus profilometry, and white light interferometry which is commonly used in our study. Real-space imaging techniques can provide a direct visual interpretation of the surface morphology. Diffraction techniques include high-resolution low-energy electron diffraction (HRLEED), reflection high-energy electron diffraction (RHEED), X-ray diffraction, and light scattering. For these techniques, surface morphology information including roughness could be obtained from the angular distribution of the diffracted pattern. They also provide non-contact measurement and are capable of getting surface information over a larger area in a short time compared with real-space imaging techniques. Condensation chemical vapor deposition20, initiated chemical vapor deposition33, plasma-enhanced chemical vapor deposition (PE-CVD)52 and other forms of VDP typically result in smooth, homogeneous films. Although iCVD technique is capable of fabricating nano-scale pattern within the vapor-deposited films10, and can be used to deposit conformal polymer coatings directly on micron-sized particles and carbon nanotubes11, these fabrication processes and iCVD in general, offer little control over film morphology and spatial heterogeneity. The introduction of non-reactive, immiscible species into a vapor deposition process has been proved effective to leverage phase separation and induce film heterogeneity. Porous or micro-structured polymers including high-glass-transition-temperature (Tg), rigid-rod polyimide has been fabricated by 14 introducing immiscible nano-sized particles. Porous films53, 54 can be fabricated by inducing thermal decomposition in these particles (porogens) that are homogeneously dispersed in a polymeric matrix via vapor deposition. The porogen molecules decompose to give small volatile fragments which either leave the polymeric matrix to form pores, or are trapped and may affect transport through chemical interaction. Various N-boc amino acids including N-boc dodecanoic acid provide potential candidates for porogens. They are stable at the evaporation temperature, but decompose while the polymer is still stable and under the Tg of the polymer matrix. The porosity and pore size are related to the weight percentage of porogen and characteristics of the polymer including molecular weight. Alternatively, nano-metal particles can be co-evaporated with polymer precursors, and polymer films with a very large variety of nanostructures can be fabricated through combination of two vacuum deposition techniques.55 Thin polymer films are deposited using plasma polymerization or traditional CVD approach, and metal particles are embedded during deposition of polymer film, mainly by simultaneous metal evaporation or metal sputtering. Metal particle distribution, size, shape and orientation can be controlled by adjusting deposition conditions including metal particle type and plasma density55, which in turn change the electrical and optical properties of deposited films. As an example, our laboratory recently co-deposited copper phthalocyanine (CuPc), a well-studied p-type organic semiconductor, with vacuum deposited polyimide to influence film morphology. Resulting films show evidence of 15 embedded CuPc particles at the surface and in the bulk of fine lateral structure with a length scale of about 100 nm.8 A similar approach has been attempted in our laboratory to deposite structured or even porous polymer films using the techniques combining iCVD and polymerization-induced phase separation (PIPS). PIPS has also been widely used to produce and control porosity in thermosetting resins.56 This method is unique because here phase separation (nucleation and coarsening) and polymerization occur simultaneously. This approach consists of polymerizing a mixture of the polymer precursors and a non-reactive solvent, termed the porogen, which will template the porosity. The porogen must be a solvent for the monomer precursor but a non-solvent for the polymer. Thus, owing to the substantial decrease in the entropy of mixing on polymerization, the initially homogeneous solution separates into a solvent-rich phase and polymer-rich phase. When polymerization and phase separation are complete, the solvent may be removed by freeze drying, leaving a porous structure,57 as illustrated in Figure 1.8. The key properties of porous materials including pore size and porosity will depend on molecular weight and crosslink density of polymer, porogen species as well as weight percentage of porogen, temperature and the amount of time the system resides at this stage. 16 Liquid one-phase system Solid-liquid two phase system Solid-gas two phase system Figure 1.8 Principle of polymerization-induced phase separation. Therefore, during the deposition run, the gas mixture containing monomer, initiator and non-reactive solvent (porogen) flows through the packed hot-zone, and initiator molecules are thermally dissociated to free radicals. The free radicals will diffuse then condense on the cool substrate and undergo free radical polymerization. During early stage of polymerization, the free energy of mixing remains favorable and whole system is homogenous. However, as the polymer chain grows, mixing becomes unfavorable. polymerization-induced It is expected that at phase separation occurs, a critical leaving chain length, polymer-rich and porogen-rich domains. As polymerization continues, polymer will become glassy and the phase-separated morphology will be “arrested”. The porogen then will be removed after the PIPS completes, leaving porous morphology. In this process, molar flow rate of each component can be easily adjusted. In principle, molecular weight, crosslink density of poly (methyl methacrylate), weight percentage of porogen can be changed as the film grows, leading a one-dimensional, precise control of film morphology in the direction of film growth. 17 1.5 Objective and outline The objective of this thesis is to determine the feasibility of controlling morphology of vapor-deposited polymer films using vapor deposition polymerization (VDP) techniques. Although several techniques have been developed successfully to make porous or structured polymer materials, however, no approach has been unanimously successful in enabling a simple, environmentally friendly and cost effective process to tune film morphology. To our knowledge, precise spatial control over polymer morphology along one dimension has not been demonstrated. Chemical vapor deposition has the ability to adjust the molar flux ratios of gaseous precursor components during deposition run therefore provides an easy and promising way to fabricate materials with precise control of morphology. In this thesis, the effect of process conditions on film morphology is explored using both step-growth and chain-growth chemical vapor deposition techniques. Non-reactive, immiscible species are introduced in the vapor deposition process to induce heterogeneity. In the following chapters, the fabrication of condensation and addition polymers, and the control of film morphology using chemical vapor deposition techniques will be discussed in detail: Rigid-rod Polybenzoxazoles (PBO) films were made using condensation chemical vapor deposition (Chapter 2). PBO precursors were vapor-deposited then thermally cured under inert gas condition. At high curing temperature (500~550°C), precursors undergo decarboxylation to form partially aromatic PBO. The successful fabrication of PBO precursors illustrates the versatility of CVD process, and provides 18 hands-on experience to achieve morphology control in the condensation polymer films. The effects of non-reactive component, i.e. copper phthalocyanine (CuPc), on morphology of vapor-deposited polyimide film were studied (Chapter 3). A new three-source vapor deposition system was custom-built to study the structure development in polyimides. Various dianhydride/diamine reactant pairs were co-evaporated and CuPc molecules were embedded into the polyimide matrix via thermal evaporation. The polyimide morphology can be adjusted by changing processing conditions including annealing temperature. An axisymmetric iCVD vacuum reactor was designed and custom built (Chapter 4). Using this reactor, smooth and homogeneous poly (methyl methcrylate) (PMMA) films can be grown from vapor feeds of methyl methacrylate (MMA) monomer and t-butyl peroxide (TBPO) initiator. We were trying to determine how this axisymmetric iCVD reactor is suited to explore depositions involving multiple components. Several key reactor operating parameters, were systematically varied to understand film growth kinetics. These include the hot-zone temperature, reactor base-pressure, substrate temperature, and the monomer/initiator molar feed ratio. A non-reactive solvent (t-butanol) vapor was then introduced into the deposition of PMMA to induce morphology change, i.e. dewetting of polymer films from substrate to form polymer droplets (Chapter 5). We found that different deposition conditions influenced whether dewetting occurred. Moreover, control over lateral length-scale was demonstrated through systematic adjustment of solvent 19 and carrier gas flowrates. An autophobic dewetting mechanism was proposed to describe the dewetting mechanism during the vapor deposition process. The next chapter begins with a discussion of vapor deposition of micron-thick PBO films and will provide a platform to study simultaneous film growth and phase separation during deposition—the topic of Chapter 3. 20 Chapter 2. Vapor Deposition and Curing of Polybenzoxazole Precursors 2.1 Introduction 2.1.1 Polybenzoxazoles (PBOs) Throughout the 1960s several new heterocyclic and aromatic polymers were developed to address the demands of the aerospace industry.58 Rigid-rod macromolecules with high glass transition temperatures and high thermal stability were developed including polyimides59, polybenzimidazoles60, polybenzoxazoles61 (PBOs) and polyquinoxalines62. PBOs in particular have been investigated over the past few decades because of their unusually high modulus and tensile strength in the fiber form, their excellent thermal stability, flame resistance, and their good hydrolytic and solvent resistance.63-67 Fully aromatic PBO fibers (with commercial name Zylon®) can be obtained by the condensation reaction of 4,6-diamino-1,3-benzenediol dihydrochloride with terephthalic acid (TA) in the presence of poly(phosphoric acid) (PPA) followed by dry-jet, wet-spinning process.63 In the dry-jet, wet-spinning process, the material is placed under high levels of tension while it is coagulated. The resulting fibers exhibit an impressive tensile strength of 5.8 GPa, an elastic modulus of 270 GPa,65 and have a characteristic degradation temperature as high as 680 °C.68 21 PBO’s properties depend on molecular weight, the degree of axial chain orientation between polymer backbones, and the level of non-covalent interactions between chains.69 Not surprisingly, there are enormous differences between the properties of fully-aromatic PBO, i.e. poly (p-phenylenebenzo-bis-benzoxazole) (1), and those of partially-aromatic PBO, such as 2 (Figure 2.1). PBO 1 is a rigid-rod macromolecule with a large perisistence length.70, 71 For PBO 1, there are few opportunities for kinks in the polymer backbone, and free torsion rotation about C-C bonds has little effect on the overall polymer configuration. On the other hand, the bonding geometry of 2 permits several kinks in the backbone due to the various rotational isomeric states about C-C bonds. These rotations improve the solubility and processibility of PBO 2 at the expense mechanical properties. N N N O O O n O N n (1) (2) Figure 2.1 Structural comparison of fully-aromatic (1) and partially-aromatic PBO(2). Laboratory synthesis of semi-aromatic PBOs, such as 2, has been developed and involves the use of heterocyclic precursor polymers.72, 73 This synthesis strategy enables the precursor polyamide to be processed, and it does not require aggressive solvents such as methanesulfonic acid or poly (phosphoric acid). For example, the reaction between pyromellitic dianhydride (PMDA) and 3,3’-dihydroxy-benzidine 22 (DHB) (Figure 2.2) results in polyhydroxyamide 3 which can be thermally cyclodehydrated to form a polyimide. Upon further thermal treatment to ~440 °C, biphenyllic hydroxyl groups react with the imide carbonyls to form new five-member, benzoxazole rings. At even higher temperature (500~550 °C) the material undergoes decarboxylation and releases carbon dioxide to form partially aromatic PBO 2.74 Attempts to achieve fully aromatic PBOs 1 using this strategy have not succeeded. Rather, direct reaction of 4,6-diaminoresorcinol with PMDA only yields the polyimide precursor and does not rearrange to form 1. In this case, thermal conversion is difficult due to the rigid structure of polyimide precursor. An alternative technique, vapor deposition polymerization, was explored as an approach to obtaining new high performance films. Over the past decade, full density, high performance polyimides17, 18, 75 , polyquinoxalines76, and poly(oxadiazoles)77 have been prepared using VDP techniques. VDP is a solventless process whereby film thickness is easily modulated, conformal coatings can be achieved down to the nanometer length scale, and resulting coatings are free of defects associated with solvent evaporation. This study is to investigate the suitability of VDP to process traditionally intractable PBOs. DHB and PMDA were co-deposited onto flat substrates to form poly (amic acid) films. Reactant fluxes from each source must be well balanced to achieve quality films. Coatings were cured under inert gas conditions, resulting in the conversion to polyimides (150-250 °C), and, at higher temperatures (550-550 °C) to semi-aromatic polybenzoxazoles. Physical and chemical changes occurring during the curing process were then studied with Fourier 23 transform infrared spectroscopy (FT-IR), thermogravimetry analysis (TGA) and nanoindentation experiments. Vapor-growth of polyimides and curing to form PBO films results in smooth, homogeneous coatings, and is the first step to achieve morphology control in vapor-deposited polyimides. HO H2N O O OH NH2 + O O O O HO O O OH H N HO H N 3 OH O n O 150-200°C -2nH2O O HO O OH N N O O -2nCO2 n 500-550°C O O N N n Figure 2.2 Synthesis of semi-aromatic PBO involving thermal rearrangement of polyimide precursor. 24 2.2 Experimental 2.2.1 Materials Pyromellitic dianhydride (PMDA) was purchased from Lancaster Chemicals (97% purity) and 3, 3’-dihydroxybenzidine (DHB) was obtained from TCI America (99% purity). Prior to coating runs, PMDA and DHB were vacuum-dried overnight at 100°C. For solution synthesis, PMDA was recrystallized twice before use according to reference78 and DHB was used without further purification. N, N-dimethylacetamide (DMAc) was purchased from Alfa Aesar and was used as received. Potassium bromide (KBr) disks (13 mm in diameter and 1 mm in thickness) were purchased from International Crystal Laboratories for FT-IR studies. 2.2.2 Vapor deposition polymerization of PBO precursor 3 All attempts to vapor-coat PBO precursors were made using a custom-built vacuum deposition chamber. This chamber has been extensively used in studies of polyimide materials and is described in detail elsewhere.79 A simplified drawing of the apparatus is shown in Figure 2.3. The chamber consists of two temperature-controlled monomer evaporators that are separated from the target substrate by 5.0 cm. The individual evaporation rates of PMDA and DHB were obtained by measuring the mass before and after deposition, to an accuracy of ± 0.1 mg using a balance. The monomer evaporation rates were measured for different evaporation temperature to determine the conditions that yield equal-molar deposition of the monomers. A retractable shutter (3× 5 cm2) is used to shield the substrate from 25 deposition until the monomer sources reach their steady-state evaporation rates. For each run, monomers were preheated to their deposition temperatures for 30 minutes prior to opening the shutter. The substrate was rotated at 20 rpm to improve film uniformity. The chamber pressure during deposition was observed to be between 210-5 and 510-5 Torr. These conditions resulted in polymer deposition rates of about 3 m / hr. For this study films were vapor-coated to about 1 m. Figure 2.3 Schematic diagram of the deposition system inside the vacuum chamber. 2.2.3 Solution synthesis of PBO precursor 3 Poly (amic acid) (PAA) 3 was synthesized from the low-temperature reaction of PMDA with DHB in the presence of N, N-dimethylacetamide. DHB (2.5 g, 11.57 mmol) and DMAc (25 g, 26.60 ml) were placed in a 100 ml three-necked flask and 26 stirred at 0 °C for 30 minutes under nitrogen purge. In a separate flask PMDA (2.52 g, 11.57 mmol) was dissolved in DMAc (25.2 g, 26.84 ml), and this solution was added to the stirring DHB solution through an additional funnel. The reaction mixture was stirred vigorously at 0 °C for 1 hour, then at room temperature for 24 hours, yielding a 10 wt % solution of poly (amic acid) 3. The resulting solution was spun-cast (3000 rpm) onto KBr optics and glass substrates and the solvent was evaporated in vacuo at 50°C for 6 hours. 2.2.4 Curing of PBO precursors As-deposited and solution-cast films were thermally cured to different temperatures (200, and 550 °C) using various heating rates (10, 1, and 0.1 °C / min) under inert gas condition (Ar). During the high-temperature cure, even small amounts of gaseous impurities (oxygen) were found to initiate thermal decomposition of films and were scrupulously avoided. 2.2.5 Characterization The thickness of VDP films was measured using a film measurement device (Filmmetrics, model: F20). FT-IR spectra were recorded using a Nicolet 20SXC infrared spectrometer. Vapor-coated films were examined using polarized optical microscopy. Thermal Gravimetric Analysis (PerkinElmer Pyris) was performed on as-deposited films at a heating rate of 10 °C/min. Mechanical properties (elastic modulus and hardness) were measured using nano-indentation techniques (MTS, Nanoindenter III). Loads were applied to the sample using a Berkovitch pyramidal 27 diamond indenter until a specified displacement (~50 nm) was reached. The sample thickness was always at least a factor of ten larger than the indenter displacement. Raw data gathered included load versus displacement curves. The hardness and elastic modulus were calculated and averaged from six different indents. For these calculations, the Poisson’s ratio was taken to be 0.41 for poly(amic acid)80, 0.33 for polyimide80 and 0.30 for poly (p-phenylenebenzo-bis-benzoxazole)81. 2.3 Results and discussion 2.3.1 Sublimation of PMDA and DHB monomers Vacuum thermogravimetric analysis (vacuum TGA) was performed to find desired evaporation temperatures for potential precursor monomers. This technique allows the vapor pressure of each tested materials to be calculated as a function of temperature. This makes it much easier to achieve stiochiometric balance of monomer fluxes during co-depositions. Figure 2.4 shows the linear relationship between logarithm of vapor pressure (log P) and the inverse of temperature (1/T) for PMDA and DHB, respectively. The vapor pressure can be calculated from this figure which allows the relative evaporation rates for any temperature to be predicted. These results can be used as a good starting point for determining evaporation temperatures of monomer sources. 28 -0.1 Log P (Pa) -0.2 -0.3 -0.4 -0.5 -0.6 DHB PMDA -0.7 2.1 2.2 2.3 1/T (1/K) 2.4 -3 2.5x10 Figure 2.4 Vapor pressure curves of PMDA and DHB from vacuum TGA measurement. Based on the preliminary results from vacuum TGA, several depositions were run to achieve a stiochiometric balance of monomer fluxes leaving the two evaporators and results are summarized in Table 2.1. The molar-loss ratio (MLR) is defined as the ratio of moles (PMDA to DHB) leaving the evaporators during a 5-hour coating run. Because molecular transport to the film depends on several factors, including reactor geometry, this ratio does not correspond directly to the molar composition of the as-deposited film.82 Nevertheless, the molar-loss ratio is a useful quantity—it is simple to measure experimentally, and it serves as a good indicator of molar flux balance. In order to achieve a MLR near 1:1, temperatures were adjusted to 213 °C and 153 °C for PMDA and DHB, respectively. 29 Table 2.1 Molar loss ratio at different operating conditions. Evaporator Temp. (°C) Molar loss ratio Run PMDA DHB (PMDA:DHB) 1 153 163 8.2 2 153 203 4.0 3 158 213 1.5 4-9 153 213 1.14 ± 0.03a a) Reported error is indicative of reproducibility and was calculated as the standard deviation of six runs. As expected, films grown under these conditions were of higher quality—they were light-yellow in color, pinhole-free, transparent, and adhered well to glass substrates. Films grown with much more PMDA (MLR>2) showed small white crystals on their surface (birefringence under polarized microscopy) and cannot stand high temperature. Upon curing, these films have more pinholes and defects than those deposited near stoichiometric balance. 2.3.2 Change of chemical structure upon curing Selected FT-IR spectra obtained before and after thermal treatment are shown in Figure 2.5. The vibration modes corresponding to relevant peaks are listed in Table 2.2 and are based on peak assignments made by others.16, 74, 83 It appears that the major component of as-deposited films is the PBO precursor, the o-hydroxy poly (amic acid), as evidenced, for example, by the intense and broad absorption band at 1650 cm-1 due to the amide C=O stretch. However, FT-IR data indicate that 30 unreacted monomers are also present. The sharp absorption at 1853 cm-1 is assigned to the symmetric carbonyl stretching mode of the dianhydride monomer, and the peak at 1503 cm-1 is partly attributed to the aromatic ring mode of free DHB. The presence of unreacted monomer was not surprising; in our prior study16 of polyimide precursors, we also identified unreacted monomers in as-deposited films. Increasing the substrate temperature during the deposition run may be an effective approach to eliminate unreacted monomers in as-deposited films. Figure 2.5 b shows an FT-IR spectrum of a PBO-precursor film following heat-treatment to 200 °C. The anhydride monomer absorptions were no longer present, and two new imide absorptions were present: at 1780 cm-1, due to the symmetric C=O imide stretch, and at 1380 cm-1, due to the imide C-N stretch. The last spectrum, shown in Figure 2.5 c, was taken of a film heated to 550 °C and held there for one hour. Medium intensity absorption bands present at 1600 cm-1 and 1460 cm-1 confirm the formation of benzoxazole rings.74, 83 The complete disappearance of imide peaks following the high-temperature curing suggests the thermal conversion from polyimide to PBO is complete. 31 Table 2.2 FT-IR peak assignments for vapor-deposited poly (amic acid), polyimide and PBO. No. Wavenumbers (cm-1) Vibrational Mode 1 1853 Symmetric carbonyl stretching of dianhydride 2 1790 Asymmetric carbonyl stretching of dianhydride 3 1780 Symmetric C=O imide stretch 4 1650 Amide C=O stretch 5 1600 Benzoxazole ring mode 6 1503 DHB aromatic ring mode 7 1460 Benzoxazole ring mode 8 1380 Imide C-N stretch 9 1241 Ether C-O-C and anhydride C-O-C stretching 32 a) 0.8 Dianhydride carbonyl stretching T 0.6 DHB ring mode 0.4 0.2 Amide C=O stretch b) T 0.6 Imide C-N stretch 0.4 0.2 Imide C=O stretch c) Benzoxazole ring mode T 0.9 0.8 2000 1800 1600 1400 -1 Wavenumber [cm ] 1200 Figure 2.5 FT-IR Spectra of (a) as-deposited film (b) film cured at 200 °C for 5 hours, and (c) film cured at 550 °C for 1 hour. The spectrum in (c) was taken using reflective mode. 33 2.3.3 Thermal properties A TGA scan of an as-deposited film (monomer loss ratio: 1.2) acquired under N2 atmosphere is shown in Figure 2.6. The sample continuously loses mass over the entire heating range, even as high as 600 °C. The continuous mass loss is believed to be associated with the evaporation of unreacted monomers. In addition to this continuous mass loss, discrete weight reductions were also observed and are associated with chemical transformations. A mass loss of about 14 % was observed between 180-220 °C. This is a little larger than the theoretical value of 8 % due to the loss of water molecules as amide linkages are converted into imide linkages. The polymer also showed significant mass loss at temperature greater than 400 °C. Part of this mass loss is attributed to decarboxylation. The fully cured PBO had a residue at 650 °C that was about 50 % of its original mass. This temperature is slightly lower than the characteristic half-decomposition temperature of commercial PBO fibers (670.7°C) in nitrogen.68 However, the thermal stability of the vapor-deposited PBO studied here far exceeds many common thermoplastics; for example, polystyrene decomposes at around 450 °C and polyimide, also considered a high-performance material, begins to decomposes at 560 °C.84 This suggests that the hydroxyamide precursor was transformed to higher char yield structures upon curing. 34 Imidization 0.9 Weight [%] 0.8 Decarboxylation 0.7 0.6 Thermal Decomposition 0.5 0.4 200 400 600 Temperature [°C] 800 Figure 2.6 TGA thermogram of as-deposited poly (amic acid) film. 2.3.4 Mechanical properties of PBO Mechanical properties are the hallmark of PBOs. Nanoindentation is ideally suited for mechanical measurement of micron-thick films because it involves extremely small volume displacements. Nanoindentation results on vapor-deposited, solution-cast, and thermally cured films are compared in Figure 2.7. Compared to solution-cast films, as-deposited and cured VDP films always exhibit higher or comparable Young’s modulus and hardness values. This result is a little unexpected since VDP films are believed to be of lower molecular weight than solution-cast films. VDP involves a solid-state polymerization that, in the absence of solvent, is kinetically hindered due to low monomer and oligomer mobility.16 However vapor-deposited films show unique properties since VDP can be regarded as a layer-by-layer process. Prior X-ray studies of vapor-deposited polyimides confirmed 35 that chain axes are preferentially oriented parallel to the surface.19 In this study, differences in Young’s modulus between VDP and solution-cast films are attributed to an overall higher degree of chain ordering and packing in the VDP form. It is difficult to assess, quantitatively, how chain anisotropy affects the Young’s modulus parallel (E) and perpendicular (E) to the surface. While nanoindentation is suitable for thin films, the measured modulus is a weighted average of E and E. All heat-treated polymers show higher Young’s modulus than as-deposited or as-cast polymers. The modulus of as-deposited films increased significantly upon converting to the polyimide, and then increased slightly upon achieving PBO. The modulus improvement is attributed to the loss of chain flexibility as hydroxyamide chains are converted to more rigid, fully-cyclized structures. PI and PBO films also exhibit higher values of measured hardness than their PAA precursors—regardless of whether they were solution-cast or vapor-deposited. 36 a) b) Figure 2.7 (a) Modulus and (b) hardness of vapor-deposited and solution-cast films at different stages of curing corresponding to poly(amic acid) (PAA), polyimide (PI), and polybenzoxazole (PBO). 37 Mechanical properties also depend on curing rate (Figure 2.8). Since vapor-processed PBO films usually cracked after curing at high temperatures for long times (>1 hour), we choose to focus on the properties of vapor-processed polyimide to understand the influence of curing rate on mechanical properties. VDP samples imidized at a slower heating rate of 0.1°C/min (longer annealing time) exhibit higher modulus values than those imidized at 1 °C/min and 10 °C/min. Films imidized at the slower rate even exhibited higher modulus than corresponding PBOs cured at 10 °C/ min. Slow curing may lead to chemical crosslinking and different mechanical properties as suggested by other authors.79 Our results agree well with the study by Chang et al. on the curing reactions of solution-cast polyamide PBO-precursors.85 They found that mechanical properties strongly depend on the curing routine. In their study, the values of the tensile strength and modulus of the precursor films were enhanced remarkably by increasing the annealing temperature and time. 38 a) b) Figure 2.8 Effect of curing rate on the mechanical properties of cured polyimide: (a) modulus and (b) hardness. As mentioned in the introduction, mechanical properties of fully and partially aromatic PBO should differ due to differences in bonding geometries. Indeed, the measured modulus of partially aromatic vapor-processed PBOs (12.5 GPa) is over twenty times lower than that of fully-aromatic Zylon® fibers (270 GPa). But the reported mechanical properties are still much better than most commodity polymers, and they exhibit outstanding thermal stability. For Zylon fibers, the alignment of molecules along the PBO fiber axis is extraordinarily high, and polymer chains become even more aligned when cured under a high stress field.64, 69 Clearly, chain 39 alignment and ordering plays a crucial role in determining the mechanical properties of PBO. Mechanical processing of PBO such as heat-treatment under uniaxial or biaxial stresses may be a strategy to improve mechanical properties of vapor-deposited PBO films. 2.4 Summary The successful fabrication of PBO shows the versatility of CVD process. We have demonstrated that VDP techniques can be applied to fabricate micron-thick PBO films. Co-deposition of PMDA and DHB monomers results in poly (amic acid) precursor, curing of as-deposited poly (amic acid) under inert gas atmosphere leads to the formation of polyimides at about 200°C and subsequently to partially-aromatic polybenzoxazoles at about 550°C. FT-IR studies confirmed the existence of unreacted monomer and the formation of polyimide and PBO upon curing. TGA studies revealed mass losses due to imidization and decarboxylation, as well as a continuous mass loss of unreacted monomer. Nanoindentation studies showed that the deposited film’s Young’s modulus and hardness can be improved by a factor of three after the conversion from poly (amic acid) to PBO, and that the curing rate is an important process parameter. Most importantly, this study demonstrates, for the first time, that high performance PBO films can be synthesized using a solventless, VDP approach. Moreover, the fabrication of homogeneous polyimide and polybenzoxazole films provides hands-on experience and is a first step toward exploring morphology 40 control in vapor-deposited polyimide films which will be discussed in the next chapter. 41 Chapter 3. Morphology of Vapor Deposited Polyimides Containing Copper Phthalocyanine 3.1 Introduction 3.1.1 Polyimides Polyimides are an important group of polymers because of their many desirable properties including excellent mechanical properties, low dielectric constant, low relative permittivity, high breakdown voltage, good processability, wear resistance, radiation resistance, inertness to solvents and long-term stability.14 Because of these characteristics, polyimides have been used in a wide range of applications including dielectric, high-temperature adhesive, photoresist, nonlinear optical material and membranes. Polyimides have also been viewed as a potential n-type semiconducting materials. Nephthalene 86 86, 87 , perylene 87 and biphenyl -derived polyimides all exhibit n-type properties in recent study, which make them perfect candidate in organic electronic devices including organic field effect transistors88, light-emitting diodes89 and photovoltaics90. The excellent mechanical, thermal and chemical properties of polyimides provide a significant advantage, since many semiconducting polymers show relatively poor stability. The synthesis of aromatic polyimide was first reported in 1908 but it was not until late 1950s that DuPont developed a successful commercial route to achieve high-molecular-weight polyimides.59 Its Kapton® brand polyimide type NH films are 42 prepared by reacting 4,4’-oxydianiline (ODA) with pyromellitic dianhydride (PMDA) in solution. The resulting poly (amic acid) precursor solution is then cast and cured at 300 °C to form the insoluble polyimide (Figure 1.3). Vapor deposition polymerization (VDP) is an alternative to the industrially important spin-coating (SC) technique to fabricate polyimide films. It was developed independently in the mid 1980s by Iijima75 et al. and Salem18 et al. respectively. Heated dianhydride and diamine monomers are co-deposited onto a solid substrate in vacuum then followed by imidization reaction. In most of the following investigation of polyimide films prepared by VDP process, PMDA and ODA were used as dianhydride and diamine components, respectively. Other diandydride and diamine precursors, such as 2,2-bis(3,4-dicarboxyphenyl) hexafluoropropane dianhydride (6FDA)91, naphthalene tetracarboxylic dianhydride (NTDA)8, 92, decamethylene diamine (DMDA)93, were also used recently. Because this process eliminates solvent, this can be advantageous in some applications, where the use of solvents can have deleterious effects or where contaminations have to be strictly avoided. It is also capable of depositing on complex nonplanar geometries such as spheres.82 3.1.2 Copper phthalocyanine Copper (II) phthalocyanine (CuPc) is a commercially available macrocyclic metal complex that possesses nonlinear optical and semiconducting properties (Figure 3.1). Together with other phthalocyanine derivatives, the chemically and thermally stable CuPc has wide applications in dye processing, spectral sensitization, chemical sensors, and optical data storage.94 43 Figure 3.1 Molecular structure of copper phthalocyanine (CuPc). The semiconducting (p-type) behavior of metal phthalocyanines was first observed in 194895 and they have since attracted great interest in advancement of prototype organic semiconductors. It is typically spun-cast or co-deposited together with a polymer onto a substrate. With nano-composites composed of mixture of non-covalently bound CuPc and polymer, one can achieve an attractive blend of the optoelectronic properties of CuPc materials with the processability of polymers. As such they are of major importance in the fabrication of photoconductive films and photovoltaic devices.55, 96 Polymer hosts are valuable platforms in these hybrid nanocomposites. Relatively polar polymers, such as poly (vinyl fluoride) and polyacrylonitrile, has already been shown to have a very significant effect on the performance of the CuPc particles by enhancing the photogeneration of charge carriers. It has been shown that the electron mobility of CuPc increases five times when it changes from granular to rodlike crystal structure in polymer matrix.97 44 Therefore, for applications including photovoltaics and organic light-emitting diodes (OLED) devices, control of thin film morphology is crucial. 3.1.3 Organic photovoltaics (OPV) Organic photovoltaic (OPV) devices have drawn great attention in the past decade and several reviews on OPV have been published.98, 99 There are many reasons for the interest in OPVs when comparing with the silicon-based photovoltaics (PV). The OPVs offer low cost, low thermal budget, solution processing, flexible substrates and a very high speed of processing. However, photovoltaic devices incorporating organic materials display low power conversions (less than 5%) compared with silicon-based PV (which are over 20 %). In OPV, excitons, which could be regarded as electron-hole pairs, are created via photoexcitation, then excitons must be dissociated in order to take advantage of the excited electrons. The earliest OPVs are Schottky-type devices in which a contact is formed at one of the organic-electrode interfaces in a metal-organic-metal sandwich (Figure 3.2 a). Such devices are inefficient as charge photogeneration only takes place in a thin layer near metal-organic interface, limiting the quantum yield of charge photogeneration. Organic pn-heterojunction is an improvement to Schottky-type devices (Figure 3.2 b). It is based on charge generation at an interface between electron donating (n-type semiconductor) and electron accepting (p-type semiconductor) materials. Then electron will travel through the n-type semiconductor and external circuit before recombining with holes at the anode. 45 a) b) Figure 3.2 Two types of OPV devices: (a) Schottky-type junction and (b) pn-heterojunction. The performance of a bilayer OPV is determined by the efficiency of charge generation and transport. One of the limitations for a two-layer pn-heterojunction OPV is that charge photogeneration only takes place in a thin layer near the organic heterojunction. Quantum efficiency of the devices will be limited because only part of incident light will be absorbed in this region. One method to increase the width of photogeneration region is to co-deposit two different organic semiconductor materials to form a bulk pn-heterojunction. In this case, electrons and holes are generated in expanded mixed layer and are swept to the transport layers by built-in chemical and electric potentials. The ability to vapor-deposit polyimide films will now be extended to fabricate bulk pn-heterojunctions by co-deposition of n-type vapor-deposited polyimide and p-type CuPc will be studied in this chapter. The effect of CuPc particles on polyimide/CuPc composite morphology will be investigated in detail. Factors including surface energy all affect morphology, making it difficult to control the bulk 46 interface geometry.100 The VDP process is shown as a viable approach toward quenching phase-separated morphologies of polyimide films containing copper phthalocyanine, therefore will be used as an ideal technique to fabricate bulk pn-heterojunction. This study builds on earlier studies involving co-deposition of CuPc with vapor grown poly(p-phenylene)101 and polyimdes102. The growth of CuPc into ordered, thin film is an important aspect of charge transport and device efficiency. CuPc was selected for this study because its crystal habit and morphology are very sensitive to processing conditions including CuPc purity, substrate type, and annealing conditions.97, 103-106 3.2 Experimental 3.2.1 Materials For polyimide coatings, pyromellitic dianhydride (PMDA, 97% purity), biphenyl tetracarboxylic dianhydride (BPDA, 97%), and xylene diamine (XDA, 99%), were purchased from Sigma Aldrich and used as received. Naphthalene tetracarboxylic dianhydride (NTDA, 95%) was purchased from Sigma Aldrich and purified by sublimation. Perylene tetracarboxylic dianhydride (PTDA, 97%) was purchased from Fluka and used as received. Oxydianiline (ODA, 98%) was purchased from Fluka and was ground with a mortar and pestle before use. All reagents were stored in a dessicator when not in use. CuPc (97%, Sigma Aldrich) was 47 used as received for morphology studies. For photovoltaic device performance studies zone-refined (>99% pure) CuPc was obtained from Eastman Kodak. 3.2.2 Substrate preparation Various substrates including glass and quartz microscope slides, polished KBr disks, and pre-patterned ITO-coated glass slides were used for film deposition. Standard 3”× 1” glass slides substrates were cleaned by soaking in KOH/ iso-propyl alcohol basebath solution for two hours, followed by rinsing and drying at 110 °C. For UV/Vis studies, 1”× 1” quartz substrates were cleaned by immersing in a 70 °C solution of one part H O , one part NH OH, and five parts water followed by rinsing 2 2 4 and drying at 110 °C for 1 hour. Polished KBr optics (13 mm diameter ×1 mm thick) were purchased from International Crystal Laboratories for FT-IR studies. 1.5” × 1.5” pre-patterned ITO-coated glass was cleaned using methanol followed by an air plasma treatment for three minutes. 3.2.3 Vapor deposition of polyimide/CuPc composites A shuttered vapor deposition system shown was designed and assembled for this work (Figure 3.3). The system consists of a 45-cm aluminum cube as the reactor. -6 A base pressure of 10 Torr was obtained using an Alcatel turbomolecular pump backed with a mechanical roughing pump. The deposition system contains two low temperature effusion sources for polyimide reagents, and one high temperature source for CuPc. The distance between substrate and sources is kept at 30 centimeters. The low temperature copper evaporators were machined to contain removable ceramic 48 P10-6 Torr Figure 3.3 Three-component VDP reactor for depositing polyimide/CuPc composite. 49 crucibles that could be weighed before and after each deposition. Using this procedure, the molar evaporation rate of polyimide reagents was roughly balanced -6 prior to depositing reported films. At 10 Torr, the mean free path of gas phase monomer species is >10 meters. Therefore intermolecular collisions are infrequent, and transport of monomers from sources to the target substrate likely occurs as individual species. The high temperature source comprises of a tantalum box source mounted on a movable platform. This source was powered by a Sorenson DCS 8-125 DC power supply and was rate-controlled using a quartz crystal monitor (QCM). Prior to film deposition, all evaporators were held at steady-state conditions for several minutes. The shutter was then opened to temporarily expose the substrate to reagent fluxes. Following deposition, all polyimide films were cured at 200 °C for one hour in a tube furnace under ambient air. 3.2.4 Characterization Thickness measurements of as-deposited films were conducted using a Zygo New View 100 white light interferometer using a 20 μm bipolar scan with a 20x objective. Fourier Transform Infrared (FT-IR) absorption studies (Brüker IFS/66) were performed on films deposited on KBr optics. Raw data were collected in percent transmittance %T and were converted to absorbance A using the relationship A[λ] = -ln(%T[λ]). A PerkinElmer Lambda 900 was used to collect UV/Vis spectra for films deposited on quartz substrate. X-ray diffraction data were collected using a Philips (X'Pert PRO MPD) X-ray diffractometer equipped with an Anton Paar TTK 450 low 50 temperature camera. For elevated temperature X-ray diffraction, the sample was mounted in a custom-built hot-stage, and samples were held at set-point temperature for two hours prior to collecting data. Transmission electron microscopy was performed using a JEOL 2000 EX instrument. Samples were prepared by depositing material onto a KBr optic, followed by careful dissolution of optic with deionized water. Isolated film fragments were then floated (< 40 nm thick) onto copper TEM grids. A TopoMetrix atomic force microscope (AFM) operated in tapping mode was employed to study surface topography of deposited films. 3.3 Results and discussion 3.3.1 VDP of polyimides Studies of polyimides and diimides based on naphthalene86, 87, perylene87, 107, 108 and biphenyl86 dianhydrides show promising n-type organic semiconducting behavior. Several dianhydride/diamine reactant pairs were co-deposited in an attempt to vapor-coat polyimides. In order to achieve stoichiometric balance between dianhydride and diamine monomer, evaporation temperatures of several monomers, including BPDA, PMDA and ODA were first estimated from vacuum TGA results (Figure 3.4), then confirmed by the readings from QCMs. 51 LogP (Pa) 0.0 -0.5 ODA PMDA BPDA -1.0 2.0 2.1 2.2 2.3 1/T (1/K) 2.4 -3 2.5x10 Figure 3.4 Vapor pressure curves of BPDA, ODA and PMDA, obtained by vacuum TGA measurement. As mentioned in the chapter 1, the monomers involved in the condensation chemical vapor deposition process require high reactivity due to the absence of solvent. Selected examples of these attempts are summarized in Table 3.1. Subsequent inspection and FT-IR analysis indicated that most dianhydride-diamine combinations resulted in little, if any, condensation reaction. Only those dianhydrides containing strained five-membered rings such as PMDA and BPDA were reactive enough to undergo polymerization. While conjugated dianhydrides containing six-membered rings such as NTDA and PTDA are commonly synthesized in solution at elevated temperatures, the mobility and reactivity of these reagents is limited in the 52 Table 3.1 Summary of typical VDP attempts to grow polyimide films. dianhydride a evap. temp. [°C] evaporator molar loss rate [mol / hr] PMDA NTDA NTDA PTDA BPDA 173 210 200 340 255 9.72 × 10-5 2.4 × 10-6 1.3 × 10-6 2.1 × 10-6 9.1 × 10-5 polymer growth rate [nm / hr] ODA XDA ODA XDA ODA 123 35 113 35 140 1.93 × 10-5 6 × 10-6 2.04 × 10-6 25.3 × 10-6 9.0 × 10-5 59 no film no film no film 90 O O O a) PMDA = O O O O O ; NTDA = O O O O O PTDA = O O ; O; O O O O O. BPDA = O H2 N evaporator molar loss rate [mol / hr] O O b) ODA = diamine b evap. temp. [°C] O NH2 ; XDA = H2N NH2 . solid state. Attempts were made to promote NTDA’s reactivity by increasing the substrate temperature; however higher substrate temperature resulted in desorption of monomer species, very low sticking coefficient and no film deposition. Careful consideration of monomer flux, stoichiometry, monomer purity, and substrate 53 temperature may enable these less reactive, six-membered ring dianhydrides to be successfully deposited, and this is the subject of a future study. A new vapor-deposited polyimide, poly(4,4-oxydiphenylene-biphenyl tetracarboxylic imide) was successfully synthesized by co-depositing BPDA and ODA precursor monomers followed by curing in air at 200 °C. The vapor deposition polymerization to form poly (amic acid) and subsequent curing to form a polyimide is shown in Figure 3.5. After several attempts, evaporator temperatures were balanced, aiming to produce homogeneous polymer films containing little to no unreacted monomer. BPDA and ODA were typically evaporated at 246°C and 126 °C, respectively, yielding a (cured) polyimide deposition rate of about 130 nm/hour as measured using white light interferometry. Resulting films were transparent and pale yellow in color. The remaining study will focus on this polyimide as a host for CuPc particles. 54 O H2N O NH2 O O + O O O O O HO NH NH OH O n O 150-200°C -2nH2O O O N N O O n Figure 3.5 Reaction of BPDA and ODA to form poly(amic acid) and subsequent curing to yield polyimide. Vapor-deposited BPDA-ODA polyimide films were studied using FT-IR spectroscopy. BPDA and ODA were simultaneously deposited and cured on KBr optics and glass slides, so that both IR spectra and film thickness could be obtained. The BDPA-ODA polyimide spectrum is shown in figure 3.6a with peaks assignments made according to prior study. 16, 109 Data confirm the formation of imide group. The -1 presence of absorption peaks at 1720 and 1380 cm correspond to the imide carbonyl stretch and the imide ring mode, respectively. Different film thicknesses (0.49, 0.46, and 1.35 μm) were measured, and the samples’ absorption coefficient, α, defined as 55 the absorbance peak height per unit thickness, was found to be nearly independent of film thickness. a) a) BPDA-ODA polyimide -1 m ] 2.0 imide C=Ostr. imide ring mode 1.0 0.0 b) b) -1 m ] 0.8 -1 m ] c) c) CuPc pyrole ring str. C=N-C C=N-C bridge bridge 0.4 0.0 1.5 polyimide + 30%CuPc PI 1.0 PI CuPc CuPc 0.5 0.0 1800 1700 1600 1500 1400 -1 1300 1200 wavenumbers (cm ) Figure 3.6 FT-IR extinction coefficients of a) cured BPDA-ODA polyimide; b) heat-treated CuPc; and c) a composite polyimide/CuPc film. 56 Table 3.2 FT-IR peak assignments for CuPc and vapor-deposited BPDA-ODA polyimide. No. Wavenumbers (cm-1) Vibrational Mode 1 1790 Asymmetric carbonyl stretching of dianhydride 2 1720 Symmetric C=O imide stretch 3 1508 C=C vibration of CuPc ring 4 1503 ODA aromatic ring mode 5 1421 Pyrrole ring’s C-C stretching 6 1380 Imide C-N stretch 7 1332 Isoindoline ring’s C-C stretching 8 1286 Isoindoline ring’s C-N vibration 9 1241 Ether C-O-C and anhydride C-O-C stretching 3.3.2 VDP of CuPc/polyimide hybrid films A series of pure CuPc films with different film thickness were deposited and studied by FT-IR. As with the BPDA-ODA polyimides, CuPc films were studied to obtain a relationship between FT-IR spectra and film thickness (figure 3.6 b). The -1 CuPc absorbance peaks used for this analysis (1286, 1332 and 1421 cm ) were selected because they did not coincide with the polyimide absorbance spectrum. The absorption coefficient of these peaks is also nearly independent of film thickness. Hybrid films were synthesized by co-evaporating CuPc together with balanced BPDA / ODA precursor monomers, then the composition of polyimide/CuPc composite was quantitatively analyzed using FT-IR. Neglecting any 57 excess volume on mixing, the bulk volume fraction of CuPc and polyimide can be independently estimated from FT-IR spectra alone using absorption coefficients of pure polyimide and CuPc films we obtained above. Based on absorption coefficient calculation, the FT-IR spectrum in Figure 3.6 c corresponds to 30% (volume fraction) CuPc embedded in a polyimide host which is in roughly agreement with the controlled readings from QCMs. However, this analysis neglects interference from neighboring peaks, and it assumes stoichiometrically balanced BPDA and ODA deposition rates. The outcome also depends on which absorption bands are selected to represent pure components. Therefore, throughout the remainder of the discussion, the composition of hybrid samples will always be referred to according to the molar flux method. UV/Vis characterization of pure CuPc, BPDA-ODA polyimide and polyimide/CuPc composite was performed to understand how optical absorption of CuPc is affected when it is doped into a host polyimide film. Figure 3.7 a compares the absorption coefficient α of pure CuPc to that of cured BPDA-ODA polyimide. It shows that polyimide absorbs strongly in the UV region (< 400 nm), while CuPc dominates the optical absorption spectrum at wavelengths above 350 nm. The absorption band of CuPc at 320 nm corresponds to B-band and the doublet absorption band in the visible region is Q-band. Gouterman’s four-orbital model theoretically describes the molecular orbitals in the phthalocyanines. It is based on the top two occupied molecular orbitals (a1u and a2u) and lowest unoccupied orbital (eg) to explain the allowed transitions in the visible-UV region. In four-orbital model, the Q-band 58 * and B-band arise from transitions (π to π transition) out of a1u and a2u into the same eg orbital. The characteristic splitting of Q-band is due to in-phase and out-of-phase coupling of transition dipoles and is referred to as Davydov splitting.104, 110 a) a) -1 [m ] 20 BPDA-ODA polyi mide 15 10 CuPc 5 0 200 b) b) 400 600 [nm] 800 16 ma x = 696 nm 14 622 12 -1 [m ] 10 CuPc 616 8 688 nm 6 6 0% CuPc in PI 30% CuPc in PI 4 2 614 0 500 600 690 700 [nm] 800 900 Figure 3.7 Optical absorption spectra of: a) pure BPDA-ODA polyimide and CuPc films; b) Q-band of CuPc and composite polyimide/CuPc films. 59 The Q-band of pure CuPc and two composite films is compared in Figure 3.7 b. The intensity of the higher energy maximum is greater than that of the lower energy maxima. This is a typical feature of the α phase of CuPc.103 In the pure CuPc film, the absorption band around 700 nm is broad and tails toward the IR region. In co-deposited films, there is less absorption in this region. This suggests there are fewer CuPc-CuPc interactions in the polymer matrix compared to the pure CuPc. Additionally, the location of the doublet peaks varies between the three films. Co-deposition causes a slight hypsochromic (blue) shift of these peaks. This indicates that lower energy photons are more readily absorbed by pure CuPc than by the hybrid materials. Effect of thermal annealing on the optical spectra of polyimide film containing 60% CuPc has also been studied. Although the relative intensity of Q-band peaks changes slightly between cured and uncured films, there is little variation in the λmax positions. This indicates curing has little effect on the shape and position of the Q-band features (data not shown). Other study has shows the peak locations changed dramatically after annealing for pure CuPc films104 which is contrary to our observation. The absence of clear peak shifts upon curing indicates that CuPc particles displays lower mobility when they are embedded into the growing polyimide matrix. Therefore, the morphology and crystal phase behavior of polyimide/CuPc composite films will also be less sensitive to heat-treatment than pure CuPc film. 60 3.3.3 Morphology of composite films The film morphology is correlated with device efficiency therefore is the focus of this study. Surface and bulk morphology of CuPc, BPDA-ODA polyimide and polyimide/CuPc composite film was studied using white light interferometry, atomic formce microscopy (AFM) and transmission electron microscopy (TEM). To study the bulk morphology, TEM micrographs of films containing 30% and 60% (volume fraction) CuPc in polyimide, both uncured and cured, are included in Figure 3.8. The contrast in the images arises from the presence of CuPc which appears dark in the micrographs. CuPc concentrated regions are clearly visible in all images. In uncured films (Figure 3.8 a, 3.8 c), CuPc-rich regions are much less defined, and CuPc appears to be spherical and to be concentrated in amorphous regions. In both films, concentration fluctuations are observed with a length scale of less than 100 nm. This length scale is significantly smaller than analogous CuPc/poly(benzobisimidazobenzophen-anthroline) blends prepared using solution techniques.111 61 Figure 3.8 TEM images of vapor deposited BPDA-ODA poly(amic acid) and cured polyimide films containing about 30 and 60 vol % CuPc. 62 After composite films were cured at 200 °C, many of the CuPc molecules were ordered into rod-like structures 60-250 nm in length. Solid-state molecular rearrangement from poly (amic acid) to polyimide appears to have enabled partial crystallization to occur. It is well known that phthalocyanine materials can exist in several crystalline polymorphs, such as α, β, χ and γ polymorphs.112 The polymorphs differ based on the tilt angle of molecules within the columns and the mutual arrangement of the columns. The stable phase is β phase, while the pure CuPc film deposited at room temperature typically consist of crystallites in α phase whose shape is similar to rods.113 Transition from α to β phase can be achieved by heating the α-CuPc films above 250 °C. In vapor-deposited composite films, low mobility of CuPc particles within the polyimide matrix results in the amorphous phase at room temperature and partial crystallization at elevated temperature. This observation indicates polymer matrix is effective to quench crystallization of CuPc particles. In addition, careful inspection of micrographs shows that most of CuPc crystals are oriented parallel to the imaging plane, with a small fraction oriented perpendicularly. The shape of these features is consistent with observations of pure CuPc films, however, these crystals exhibit a slightly smaller length scale.103, 104 Lacking of mobility for CuPc particles may attribute to this difference. An alternate explanation for the smaller crystal size is that crystal growth is constrained by the extremely thin film geometry (about 35 nm for TEM measurement). As expected, cured films containing 60% CuPc show significantly more of rod-like features than films containing 30% CuPc. Dark, amorphous regions that look 63 very similar to the uncured films are still present in the cured images. The presence of these non-crystalline CuPc domains after curing suggest that much of the CuPc remains dispersed throughout the polymer. This supports the idea that CuPc has a lower mobility when supported within a polyimide host. Surface morphology of BPDA-ODA polyimide, hybrid films, and pure CuPc was characterized using atomic force microscopy (AFM) and white light interferometry. The surface of cured BPDA-ODA polyimide is smooth and featureless, with average root-mean-square (RMS) roughness around 1 nm which is less than one percent of the film thickness (Figure 3.9 a). Water is produced as a byproduct during curing, and leaves through diffusion, and this does not appear to perturb the surface. Composite film containing 30% of CuPc was also studied using AFM (Figure 3.9 b). Although it appears featureless under white light interferometry, hemispherical bump-like features with size approximately 150 nm in diameter were observed in AFM micrographs. Film containing 60% of CuPc has similar morphology with features about 50~100 nm (Figure 3.9 c). This rough morphology leads to RMS roughness increase to 22 and 14 nm for composite films containing 30 % and 60 % CuPc, respectively. The bump length scale appears to increase with film thickness, which may reflect that surface roughening is nucleated by the subsurface formation of crystalline CuPc domains. Note that the surface morphology of composite films is qualitatively different from that of pure, vapor-deposited CuPc (Figure 3.9 d). 64 a) b) c) 65 d) Figure 3.9 Atomic force micrographs of a) BPDA-ODA polyimide, b) 30 % CuPc in polyimide, c) 60 % CuPc in polyimide, and d) pure CuPc films. Film thicknesses for a-d) were measured to be 400, 264, 80, and 141 nm, respectively. Temperature dependent wide-angle X-ray scattering (WAXS) measurement was conducted to further investigate how polyimide matrix affects the stability and polymorphic behavior of CuPc (Figure 3.10). It is showed that crystal structure of vacuum-evaporated CuPc is monoclinic and CuPc macrocycles lie parallel to the substrate plane114, 115 which is in good agreement with our observations from TEM. As-deposited poly(amic-acid)/CuPc composites show diffraction with very weak scattered intensity which indicates CuPc remains in amorphous region in as-deposited films (data not shown). However, both pure CuPc and polyimide-CuPc composites exhibit Bragg diffraction peaks around 2θ = 6.8° (1.29 nm spacing) after curing, corresponding to the α-phase of CuPc.103, 105, 106 For composite films, the intensity of this diffraction peak is disproportionally weaker which suggests that the co-deposition 66 of polyimide quenches the crystallization of CuPc particles, therefore, there is still a significant amount of CuPc remains dispersed throughout the polyimide matrix in non-crystalline form. This explanation is in qualitative agreement with TEM images (Figure 3.8) that show a lower volume fraction of CuPc crystals than one would expect based on the materials’ composition. Upon heat treatment, the scattered intensity of pure CuPc peak begins to decrease around 300°C. However, the scattered intensity from the CuPc polyimide composite remains nearly constant and scattering is observed even to 350 °C. In deposited CuPc films, the α-phase is known to be metastable103, 106, 116, and, above 250 °C, the crystal transforms to the β-phase. Hence, the data presented here indicate that vapor deposited polyimide can restrain the polymorphic change and stabilizes the α-phase of CuPc to temperatures above 300 °C. 67 a) a) Pure CuPc Intensity [a.u.] 300 °C 250 °C 200 °C 6 7 b) b) 2 8 9 Intensity [a.u.] 60% CuPc in BPDA-ODA 350 °C 300 °C 250 °C 200 °C 6 7 2 8 9 Figure 3.10 Temperature dependent X-ray diffraction spectra of vapor-deposited films: a) pure CuPc, and b) 60 vol % CuPc in BPDA-ODA polyimide. 68 3.4 Summary In this chapter, vapor deposition polymerization (VDP) was coupled with phase separation and crystallization to grow novel polyimide/CuPc composite films. Various combinations of dianhydride/diamine reactant pairs were evaporated. It was discovered that monomer reactivity is important in solid-state reaction and BPDA and ODA were chosen as polyimide precursor monomers. Molar flux of each monomer was then adjusted to achieve the stoichiometric balance. FT-IR and molar flux analysis were used to independently estimate film composition, and they roughly agree. Most importantly, we have demonstrated that condensation VDP can be used to change morphology of as-deposited films effectively. Third-component, non-reactive CuPc particles were co-deposited with polyimide precursor monomers. Resulting films show evidence at the surface and in the bulk of fine lateral structure with a length scale of about 100 nm from AFM and TEM measurements, respectively. VDP technique is also effective to quench crystallization of CuPc particles. Embedded CuPc particles initially appears amorphous, as evidenced by the less-defined CuPc regions in TEM micrographs, and undergoes partial crystallization upon thermal treatment from poly (amic acid) to polyimide at 200 °C. Crystallized CuPc particles inside polyimide matrix display rod-like morphology which is the characteristic of α-phase CuPc crystallites. The morphology can be adjusted by changing CuPc volume fraction and curing temperature. Future studies will examine whether the stable morphology and high level of interfacial surface area observed in composite films 69 may be useful in developing these or other n-type polyimides as materials for photovoltaic or OLED devices. In this study, the length scale of CuPc domain is in the nanometer range. In the next two chapters, we will introduce an approach to change and control as-deposited film morphology over a larger lateral length scale. A non-reactive, immiscible solvent will be used to drive phase separation during initiated chemical vapor deposition (iCVD) of chain-growth polymers. 70 Chapter 4. Multi-component Vapor Deposition Polymerization of Poly (Methyl Methacrylate) in an Axisymmetric Vacuum Reactor 4.1 Introduction 4.1.1 Materials and application Poly(methyl methacrylate) (PMMA) (Figure 4.1) is a kind of widely studied, optically clear, glassy polymer with several thin film applications including protective coatings117, bonding adhesives117 and low dielectric layers118, 119. In its neat form, PMMA exhibits a glass transition temperature (Tg) around 105°C, well above room temperature, but when it is solvated the glass transition temperature drops to around room temperature. Generally, it is synthesized by solution free radical polymerization between methyl methacrylate monomer and free radical initiator such as t-butyl peroxide, but emulsion polymerization and bulk polymerization are also routinely used in industry and their reaction kinetics are well known. H2 CH3 C C n C O O CH3 Figure 4.1 Molecular structure of poly (methyl methacrylate) (PMMA). 71 Another important application for PMMA is to fabricate porous membranes. Torkelson’s work in 1990 demonstrated that asymmetric porous membranes could be made from PMMA (with weight-average molecular weight 93K Daltons)/porogen system using thermal-induced phase separation (TIPS) technique.120, 121 TIPS involves rapid quenching of a polymer solution to a temperature where the solvent is transformed into a non-solvent, then spinodal decomposition occurs, resulting in glassy, two-phase microstructure. Sulfolane, t-butanol or xylene have been used as porogen to introduce porosity into PMMA films. The pore size is in the micron range and can be controlled by adjusting the weight percentage of porogen, quenching temperature and time. Polymerization-induced phase separation (PIPS) is another common strategy to form two-phase, structured polymers that have been used to create interpenetrating networks122 and toughened amorphous glasses123. 4.1.2 iCVD reactor design Geometry of iCVD reactor is important to produce desirable polymer thin films. Figure 4.2 shows the setup of a traditional pancake-shape initiated chemical vapor deposition (iCVD) reactor which was custom-designed by Gleason’s group in late 1990’s.124 It has internal dimensions of 25.4 cm diameter and 3.2 cm height, used for deposition on 4-inch-diameter silicon substrates. It involved horizontal flow of monomer and initiator gases across a heated-filament array which is spaced 1.5 cm apart and positioned horizontally over a cooled target substrate at a distance of 2.5 cm. Thermal decomposition of initiator molecules into free radicals occurs near the hot-filament array. Generated free radicals and 72 unreacted monomer molecules adsorb onto the cooled substrate and irreversibly undergo free radical polymerization. Recent studies indicate that polymerization inside this iCVD reactor is limited by monomer adsorption and, within the adsorbed layer, reaction kinetics are analogous to bulk-phase free radical polymerization.35, 124 The horizontal flow of the initiator vapor eliminates the “shadowing” fact from filament array during the deposition, and at the same time, continuously provides free radicals along gas flow direction to ensure the formation of uniform film. Because of low thermal mass of filaments, high temperature (> 400 °C) can be easily achieved with very low power input (< 3 W) which minimizes the heat radiation to the cooled substrate. However, it is relatively difficult to measure the filament temperature accurately using thermocouples with comparable thermal mass, hot spot may exist and thermal decomposition of monomer or initiator species could occur during deposition process. In addition, our goal is to control the morphology of vapor-deposited polymer films by introducing non-reactive, immiscible component. However, multi-component deposition has not been reported so far using this pancake-shape design. Therefore, new reactor configuration needs to be developed to explore depositions involving multiple components. 73 Figure 4.2 A schematic drawing of the pancake-shape iCVD reactor setup for coating on flat substrates. This figure was reproduced from reference 124. In this study, we employ an alternate reactor configuration to perform initiated chemical vapor deposition process that avoids filament array altogether (Figure 4.3). First, gas precursors are passed vertically through a tubular packed-bed hot-zone which has big thermal mass. The purpose of the hot-zone is to activate free radical initiator molecules. The hot-zone is isothermal, eliminating hot-spots that could result in undesirable thermal degradation of initiator or monomer species. Following the hot-zone, the gas mixture is perpendicularly directed at a cooled target substrate, and reactants condense and polymerize on the substrate, analogous to the hot-filament process. Unreacted gases follow axisymmetric streamlines around substrate and then exit the reactor to the vacuum pump. 74 Initiator (I) Crosslinker (X) Monomer (M) Heated Hot-zone (~200°C) Solvent (S) Controllable pressure (1-20 Torr) Back-cooled substrate (-15°C) Figure 4.3 Schematic drawing of axisymmetric iCVD setup. Figure 4.4 is a drawing of the axisymmetric iCVD reactor we custom-designed and used in this study. The reactor consists of a 27.90 cm diameter aluminum base-plate that supports a glass cylinder, 27.90 cm in length, with an aluminum top. Vacuum is achieved using a mechanical pump (BOC Edwards RV8) through a pump-out port on the reactor, positioned opposite to the feed port. Reactor pressure is controlled using a downstream throttle valve (MKS Instruments) together with a Baratron capacitance manometer (MKS Instruments). Up to four different reagent gases can be supplied from separate bottles on a manifold on the up-stream side of the reactor. Reagent gases can be individually heated to ensure that supply-side vapor pressures are high enough to sustain steady flow. Reagent flowrates are regulated, prior to mixing, using separate mass-flow controllers or calibrated needle valves. After mixing the reagents 75 outside of the reactor, they are fed into the reactor through a heated transfer line (Heaters Controller and Sensors, Ontario, Canada). Upon entering the reactor, reagent gases are fed into a hot-zone consisting of an aluminum cylinder tube (ID = 11.43 cm). A band heater, combined with a temperature controller and read-out (Omega CN 9000A), was used to regulate the hot-zone temperature. The distance between substrate and hot-zone was kept at 7.62 cm in this study. The hot-zone can be packed with packing material to improve heat transfer to the supply gases. A porous aluminum gas diffuser, placed at the end of the hot-zone, is used to improve the outlet gas-flow uniformity. The distance between the hot-zone and the substrate is adjustable. The substrate can be glass slides or wafers up to 18 cm in diameter. The substrate temperature is controlled by backside cooling using a recirculating chiller/heater unit (Neslab RTE 740). The substrate temperature is also influenced by radiant heat transfer from the hot-zone and, to a lesser extent, by heat transfer from reagent gases. During operation, the substrate temperature, measured using a hand-held IR thermometer, was found to be about 5 °C warmer than the chiller temperature. The finite volume (FV) computational fluid dynamics (CFD) software Fluent was used to simulate the transport of gas species in current iCVD reactor design (Data not shown). The results indicate the gas molecules can be transported to cooled substrate uniformly. Therefore, this flow configuration was chosen to minimize mass transfer resistance to the substrate, to further decouple initiation and polymerization, and to improve film uniformity. 76 a) b) Figure 4.4 (a) Top-view of 11-inch aluminum base-plate; (b) schematic of custom-built, axisymmetric iCVD apparatus. 77 As a model system, glassy films of poly (methyl methacrylate) (PMMA) are grown using this new reactor configuration. The aim of the present study is to understand how reactor operating parameters are related to film growth rates and polymerization characteristics. Key reactor operating parameters were systematically varied. These include the hot-zone temperature, reactor base-pressure, substrate temperature and the monomer/initiator molar feed ratio. A second objective is to demonstrate how this reactor design is suited to explore depositions involving multiple components. Multi-component iCVD is demonstrated by co-condensing t-butanol solvent vapor during the growth of PMMA films. Since t-butanol is non-reactive and has a relatively high vapor pressure, it can be easily removed following deposition. Here we discuss how the presence of t-butanol affects the molecular weight of as-deposited PMMA films. This result is the first step toward developing iCVD as a technique to survey porous and structured polymer thin films with tunable morphologies. 4.2 Experimental 4.2.1 Materials Methyl methacrylate (MMA) monomer (Alpha Aesar, 99% purity), t-butyl peroxide (Sigma Aldrich, 97% purity), and t-butanol (J. T. Baker, 99.99 % purity) were all used as received, without further purification. 3” Silicon wafers (7.62 cm diameter) or glass slides were used as the substrates for deposition runs. Wafers (Silicon International) were used as received. Glass slides were cleaned by 78 soaking in a base-bath and then were vacuum-dried for overnight before each deposition. 4.2.2 Deposition of PMMA films During a typical deposition, the system was first pumped down to the operating pressure (e.g. 7 Torr) while the hot-zone was heated to the required temperature (200 °C). Next, the substrate was cooled and held at a lower temperature Tsub (-15 °C). After allowing an hour for the system to approach thermal equilibrium, monomer and initiator were introduced into the chamber at set flow rates. For all experiments, the monomer flow rate was fixed at 4 sccm and monomer/initiator feed molar ratio was adjusted by changing the flow rate of initiator. Methyl methacrylate (MMA) monomer and t-butyl peroxide (TBPO) initiator are both volatile and have relatively high vapor pressure (40.02 Torr for MMA125 and 31.34 Torr for TBPO126 @ 25°C, respectively), therefore, no heating is needed to vaporize monomer and initiator species. After the deposition was complete, the chiller and mass flow controllers were turned off. The reactor chamber was pumped down for an additional half-hour to ensure complete removal of unreacted monomers. 79 Table 4.1 Deposition conditions of iCVD experiments. a Hot-zone Reactor Substrate temperature pressure temperature Thot [°C] [Torr] Tsub [°C] 220 7 -15 Flow rate of TBPO initiator [sccm] 1 Experimental run sets base-case Hot-zone configurationb A2 A A1, A2, A3 220 7 -15 1 B A2 110, 150, 7 -15 1 0.1, 0.5, -15 1 -15, -10, -5, 1 180, 200, 220 C A2 220 1, 3, 5, 7, 10, 15 D A2 220 7 0, 5, 10, 15 E A2 220 7 -15 1, 2 a) Flow rate of monomer is kept at 4 sccm, the distance between hot-zone and substrate is 7.62 cm; b) See Figure 4.5. 80 11.40 cm A1 1.27 cm 11.40 cm A2 1.27 cm 11.40 cm A3 5.08 cm Figure 4.5 Hot-zone configurations employed in iCVD experiments. Table 4.1 lists the experimental conditions of 21 different experimental runs, and Figure 4.5 indicates the different types of reactor hot-zones used. Hot-zones differ in cylinder height; A2 and A3 were packed with glass beads (6 mm diameter) to promote gas mixing and heat transfer. Other than base-case conditions, every experiment was run only once and in the order listed in Table 4.1. Each set of experiments was designed to study different process variables: hot-zone configuration, hot-zone temperature, reactor base-pressure, substrate temperature and initiator flowrate. 4.2.3 Characterization of iCVD PMMA films Fourier transform infrared spectroscopy (FT-IR, Brüker IFS/66) was used to confirm the structure and composition of as-deposited films. Spectra were acquired 81 at 4 cm-1 resolution over the range of 650-4000 cm-1 and averaged over 16 scans. Thermal Analysis (PerkinElmer, Pyris) was performed on as-deposited films at a heating rate of 10 °C/min under nitrogen purge. White light interferometry (Zygo New View 100) was used to observe the film surface morphology and to measure film thickness and deposition rates. The thickness was averaged from measurements of three different substrate positions. Film thickness results from interferometry measurement were confirmed by an optical film measurement device (Filmmetrics, model: F20). The molecular weight of as-deposited films was determined by using an Agilent 1100 HPLC system equipped with a Viscotek triple detector array. For each sample, the film was dissolved from the substrate using N-Methylpyrrolidone (NMP, Sigma Aldrich). Polymer solutions (1 mg/ml) were prepared and filtered prior to injecting 100µl of the solution into the GPC system, containing three columns (two Viscotek Mixed Bed Low MW i-series and one mixed bed medium MW i-series column) maintained at 60 °C. The software (OmniSEC v.4.0, Viscotek) calculated the number average molecular weight Mn and weight-average molecular weight Mw by integration area of GPC traces. Results were calibrated using a set of narrow poly (methyl methacrylate) standards of known molecular weight and molecular weight distribution. 4.3 Results and discussion 4.3.1 iCVD of PMMA films using base-case conditions Several PMMA films were successfully deposited from methyl methacrylate (MMA) monomer and tert-butyl peroxide (TBPO) as an initiator 82 using the base-case conditions listed in Table 4.1. Typical thicknesses of as-deposited films were about 3 µm after 8-hour depositions. The resulting film showed consistent morphology and thickness (thickness deviation of five as-deposited films is 8%) which illustrates the reproducibility of current setup. Film thickness measurements on a 3-inch-diameter silicon wafer show good uniformity with deviation only 2 % over 28 different positions (Figure 4.6). Resulting films exhibit relatively smooth, featureless surfaces from scanning electron microscopy (SEM) measurements (Figure 4.7). The RMS roughness, as determined by white light interferometry, is around 20 nm (data not shown) which is an order of magnitude larger than the resolution of the instrument. Films are easily dissolved from substrate using a good solvent for PMMA such as tetrahydrofuran (THF). From thermal analysis, the as-deposited polymer shows a glass transition temperature (Tg) of 105 °C and undergoes thermal decomposition at around 275 °C (data not shown). These thermal characteristics agree well with PMMA standards.127 83 Figure 4.6 Thickness profile of as-deposited PMMA film (8-hour deposition) on a 3-inch-diameter silicon wafer (unit: nanometer). Figure 4.7 SEM image of as-deposited PMMA film. 84 3.0 2.5 C=O stretch C-H bend C-O stretch A 2.0 1.5 C-H stretch 1.0 0.5 0.0 3000 2500 2000 1500 Wavenumbers (cm 1000 -1 ) Figure 4.8 FT-IR spectrum of PMMA deposited using axisymmetric iCVD reactor. Figure 4.8 shows an FT-IR spectrum of as-deposited film. The film exhibits characteristic absorption bands expected for PMMA: C-O stretching (1240 cm-1), C-H bending (1452 cm-1), C=O stretching (1730 cm-1) and C-H stretching (2990 cm-1). The spectrum shows no evidence of alkene groups at 1646 cm-1 indicating that all condensed monomers either reacted to form PMMA film or evaporated and were removed from the film following deposition. Using base-case operating conditions, the number-average molecular weight (Mn) of as-deposited polymer was measured to be 25,000 Daltons. This molecular weight is comparable to the molecular weight of as-deposited polymer using the hot-filament reactor designs.27 The polydispersity index (PDI) is around 1.4 which is relatively low compared to conventional radical polymerization in 85 solution.128 The low polydispersity in iCVD process is attributed to bulk free radical polymerization in the absence of solvent, which limits chain-transfer. In addition, the mobility of polymer chains decreases with increasing molecular weight. As a result, short polymer chains grow faster than long polymer chains, leading to a slightly lower molecular weight distribution. The growth of glassy PMMA films reported here can be compared to a recent study by Chan and Gleason resulting in hot-filament iCVD growth of PMMA. In Chan’s study, polymer films could not be grown using tert-butyl peroxide (TBPO) as the initiator. It is possible that low molecular weight polymer was deposited, but no material remained after vacuum drying. However, high molar mass PMMA films were grown (~ 20 nm/min) using triethylamine (TEA) as an initiator.29 TEA requires much higher temperatures for dissociation into free radicals, and, to achieve this, the hot filament in the iCVD reactor was heated to 550 °C. The authors deduced that a higher filament temperature is necessary to support the propagation kinetics of the monomer. In the current study, high molecular weight PMMA thin films can be grown using TBPO and a relatively low hot-zone temperature of 220 °C. This finding suggests that either the hot-zone is more effective in generating free radicals, or that generated radicals are more efficiently delivered to the target substrate due to the reactor geometry and the low substrate temperature. iCVD of PMMA using the axisymmetric reactor can be broken into three steps: 1) thermal initiation inside the hot-zone; 2) monomer and free radical mass transport from the hot-zone to the substrate; and 3) reagent condensation and polymerization that occurs on the substrate (Figure 4.9). Experiments (sets A-E in 86 Table 4.1) were designed to study each of these steps. The results led to the development of simple physical models that describe, qualitatively, the physics occurring in each reactor region. 1) 2) 3) Figure 4.9 Three steps in iCVD of PMMA process. 1) Thermal initiation; 2) free radical and monomer transport; 3) condensation and reaction. 4.3.2 Hot zone configuration and temperature To better understand the efficiency of the hot-zone in activating free-radical initiator molecules, three different hot-zone designs (Figure 4.5) were implemented as shown in Figure 4.10. Comparing deposition rates from an unpacked (A1) to a packed (A2) hot-zone shows that packing with glass beads significantly increased the deposition rate, by over a factor of two. Although the residence time is significantly lower for the packed hot-zone A2 (0.25 s) compared to hollow hot-zone A1 (0.7 s), the glass spheres greatly increase the 87 thermal contact area, promoting free-radical formation. The highest deposition rate of 570 nm/hr was achieved for configuration A3 involving a packed hot-zone with an extended length. In comparing A2 to A3, the reactor length was extended by a factor of four, but the deposition rate increased less than a factor of two. This suggests that configuration A3 is approaching the equilibrium conversion of initiator to free radicals at the outlet of the hot-zone. Deposition rate (nm/hr) 600 500 400 300 200 100 0 A1 A2 A3 Figure 4.10 PMMA deposition rates for different reactor hot-zones in axisymmetric iCVD reactor operating under base-case conditions. To further study thermal initiation in the iCVD reactor, PMMA coatings were deposited at different hot-zone temperatures using reactor configuration A2. Other process conditions were held constant and are summarized in Table 4.1. As shown in Figure 4.11, the measured film deposition rate increases about an order of magnitude with increasing hot-zone temperature. Data can be fitted using an Arrhenius relationship, and the resulting slope indicates an apparent activation energy of 97 kJ/mol. This value is significantly less than the literature reported tert-butyl peroxide activation energy of 153 kJ/mol.129 This difference is attributed 88 to three factors: 1) initiator molecules never achieve thermal equilibrium within the hot-zone, 2) some recombination of free radicals may occur in the volume between the hot-zone and the substrate, and 3) additional recombination of primary radicals may occur on the target substrate. To summarize, the measured deposition rate is indirectly related to the rate of initiator dissociation. While homolytic cleavage of the TBPO peroxy bonds occurs within the hot-zone, only a fraction of the resulting primary free radicals adsorb on the target substrate and contribute to film growth. It is not surprising that many generated free radicals are lost. Compared to solution polymerization, the initiator-to-monomer molar feed ratio (1:4) in iCVD process is very high, and consequently, some recombination of gas and surface primary free radicals is expected. Inasmuch as primary radicals can undergo recombination, they can also terminate growing polymer chains. These events can influence the polymerization kinetics in iCVD films.124 89 Thot 220 200 (°C) 180 160 2.2 2.3 140 120 3 Deposition rate (nm/hr) 2 100 9 8 7 6 5 4 2.0 2.1 2.4 3 2.5 2.6 -1 (10 K ) Figure 4.11 Logarithmic plot of PMMA deposition rate versus hot-zone temperature. 1/Thot This study introduces the packed hot-zone as an alternative to the resistively-heated hot-filament free radical source traditionally used in iCVD processing. Thermal control of the hot-zone is simple and precise. Compared to the hot-filament, the packed hot-zone has a far greater thermal mass and eliminates temperature gradients that are suspected around filament edges. The elimination of hot spots may be an important factor when considering undesirable high temperature side-reactions. For the experimental conditions considered here, the kinetics of initiator dissociation within the hot-zone appear to be a key rate-limiting step for PMMA deposition. Interestingly, this observation is in contrast to a recent hot-filament iCVD study of poly(alkyl acrylates)35, 124 where 90 the deposition rate was found to depend weakly on hot-filament temperature, with activation energy of only ~ 12 kJ/mol. 4.3.3 Reactor base pressure Figure 4.12 shows the effect of reactor base pressure on measured deposition rates. The data show a maximum deposition rate (325nm/hr) at pressures about 7 Torr. The maximum can be explained on the basis of the reactor residence time, which is directly proportional to pressure. At low pressures, gases quickly pass through the hot-zone, and the extent of primary radical generation is low. At high pressures, gases pass slowly through the hot-zone, leading to higher levels of initiation. However, due to lower gas velocities, recombination of free radicals becomes significant in the volume between the hot-zone and the substrate. 350 Deposition rate (nm/hr) 300 250 200 150 100 50 0 0 5 10 Pressure (Torr) 15 20 Figure 4.12 Experimental results (○) and model fit (dashed line) illustrating the relationship between deposition rate and reactor base pressure. 91 A crude and simplified plug flow model illustrates these effects and explains, qualitatively, the rise and fall in deposition rate. This model was used to generate the least-squares fit shown in Figure 4.12. In this model, flow through the iCVD system is divided into two isothermal tubular reactors: a hot-zone (HZ) and a recombination-zone (RZ) reactor (Figure 4.13). A gas stream containing initiator molecules (I2) is fed to the hot-zone (HZ) where thermal initiation occurs. Recombination is only permitted in the recombination-zone (RZ), and the polymer deposition rate is taken as proportional to the number of generated free radicals that pass through the RZ without undergoing recombination. v0, CA0 aX HZ LHZ RZ LRZ Cdiss Figure 4.13 Simplified plug-flow reactor diagram involving one reactor for thermal initiation and a second reactor for recombination. In the hot-zone, initiator molecules are thermally activated, leading to the formation of primary free radicals: 92 I2 2I ·, [4.1] where I2 refers to initiator molecules, and I denotes primary free radicals. The reverse reaction occurs in the recombination zone. Since initiation is a unimolecular process, the reaction is taken as first order ri k i Ci , [4.2] where Ci is the gas concentration of I2, and ki is the initiation rate constant. The plug flow design equation relating the hot-zone length LHZ to the degree of initiator conversion Xi is LHZ Ci 0 v0, HZ ax XI dX i , ri 0 [4.3] where Ci0 is the initial inlet initiator concentration, v0,HZ is the total volumetric flow rate to the hot-zone, and ax is the reactor cross-sectional area. Since initiation creates additional gas molecules, the concentration of I2 decreases with conversion as 1 X i C i C i 0 1 X i . [4.4] Combining equations [4.2] – [4.4], and integrating, results in LHZ v0, HZ ki a x [ X i 2 ln(1 X i )] . [4.5] This equation relates the hot-zone reactor length to the chemical conversion. A similar procedure is repeated for the recombination reaction which is described using a second-order rate law: ri k r Ci2 [4.6] 93 where kr is the recombination rate constant, and Ci is the concentration of free radical species. For the recombination zone, if the inlet concentration of free radicals Ci,in is taken as the product, Ci0Xi, this results in LRZ v0, HZ 1 X i 1 ln(1 X r ) 1 / 4 X r X r X 4 ( 1 ) r k r a x Ci 0 2 [4.7] relating the length of the recombination zone, LRZ , to the fraction of generated free radicals that undergo recombination, Xr. Since all experiments were conducted at a fixed molar feed rate, Ci0 can be calculated using the ideal gas law. Equations [4.5] and [4.7] then relate operating pressure to the outlet concentration of primary free radicals CI·, out ~ Ci0 Xi (1-Xr). Again, this quantity is assumed to be proportional to the polymer deposition rate. The dashed line in Figure 4.12 represents a least squares fit to experimental data using this simplified plug-flow model. Parameters include two reaction constants (ki, kr), three reactor dimensions (LHZ, LRZ, and ax) and a proportionality constant, that relates the deposition rate to the outlet concentration of primary free radicals. The reactor dimensions are known (LHZ = 1.26 cm, LRZ = 6.0 cm, ax = 102.6 cm2), the initial initiator concentration is calculated to be Ci0 = × mol/cm3, and ki is around 10-5 s-1 which was estimated from the literature130, leaving only two unknowns (kr and ×) to be fitted. Note that the fitted value of kr (7.03 108 L/mols) is in rough agreement with measurements of small radical recombination rates.131 The quality of fit is reasonable and suggests that, at low pressure, deposition rate is 94 determined by thermal initiation, and, at high pressure, recombination cannot be neglected. 4.3.4 Substrate temperature The measured polymer deposition rate also depends on the substrate temperature Tsub. A semi-logarithmic plot of deposition rate versus 1/Tsub is shown in Figure 4.14. The data are nearly linear, and a least-squares fit corresponds to an activation energy of -12 kJ/mol. On the one hand, a negative activation energy is expected because lower substrate temperatures lead to higher levels of monomer adsorption, therefore faster polymerization. Such adsorption-limited kinetics were observed in analogous hot-filament reactor studies35; though, a much higher activation energy was reported (-79.4 kJ/mol). 285 280 Tsub(K) 275 270 265 260 Deposition rate( nm/hr) 4 3 2x10 2 3.5 3.6 1/Tsub 3.7 3 -1 10 (K ) 3.8 Figure 4.14 Plot showing deposition rate dependence on substrate temperature. 95 In the present study, diffusive transport of primary free radicals is also believed to limit deposition. In all experiments inertial forces are on the same scale as viscous forces. For example, the dimensionless Reynolds number, reflecting the ratio of inertial forces to viscous forces, is about 0.7 for the base-case condition (calculated using the Kinetic Theory of Gases at a temperature of 400 K). This suggests that diffusive phenomena play important roles in mass, momentum, and thermal transport and that there are no boundary layers. Furthermore, the Kinetic Theory of Gases provides scaling expressions for diffusion coefficients. In particular, the mass diffusion coefficient scales as T3/2. A lower substrate temperature can therefore reduce the gas phase temperature and the diffusive flux of free radicals to the substrate. Opposite to this effect, a lower substrate temperature increases monomer adsorption, promoting polymerization and film deposition.124 A balance of these two effects determines the observed deposition rate and explains the reduced sensitivity to substrate temperature reported here. 4.3.5 Monomer/initiator molar feed ratio Size exclusion chromatography was used to assess the molecular weight of several deposited films. Typically films exhibit molecular weights of about 20 kg/mol. The molecular weight of as-deposited PMMA polymer could be adjusted using two methods. By changing the monomer/initiator feed molar ratio from 2: 1 to 4:1, the number average molecular weight increased from 20 kg/mol to 25 kg/mol (Fig. 4.15 a). A high concentration of free radicals may terminate growing polymer chains, resulting in the low molecular weight polymer. The effects of 96 introducing a non-reactive, third component into the feed gases is shown in Figure 4.15 b. By introducing the t-butanol into the system, the molecular weight of as-deposited film increases from 20 to 32 kg/mol. Since iCVD reaction involves bulk polymerization, as chains grow longer, they lose mobility. The introduction of t-butanol is believed to improve the mobility of free radical end-groups, acting as a solvent. a) Refractive Index [M]:[I] = 4:1 Mn : ~25 Kg/mol [M]:[I] = 2:1 Mn : ~20 Kg/mol 16 18 20 22 Retention Volume (ml) 24 b) [M]:[I]:[P]=2:1:0 Mn : ~20 Kg/mol Refractive Index [M]:[I]:[P]=2:1:10 Mn : ~32 Kg/mol 12 14 16 18 20 22 Retention Volume (ml) 24 Figure 4.15 Size exclusion chromatograms of vapor deposited PMMA: a) effect of monomer/initiator molar feed ratio, and b) effect of non-reactive third component, t-butanol. 97 4.3.6 Reactor modifications In thin film deposition, one of the most important features is the film thickness. As such, it is very important to know the rate that the material, in this case PMMA, is being applied to the target substrate. However, the highest deposition rate we can achieved using our hot-zone design is 560 nm/hr which is only about half of the deposition rate obtained from hot-filament reactor.29 In our investigation of substrate temperature, we have demonstrated that diffusive transport of free radical molecules is one of the limiting steps in our iCVD process. Reynolds number Re, which reflects the ratio of inertial forces to viscous forces, is defined as: Re v z D [4.8] where ρ is the density of the gas, vz is the gas velocity, D is the diameter and µ is the dynamic viscosity. High dimensionless Reynolds number indicates greater inertial forces which allow the free radicals to flow closer to the substrate, decrease the diffusion length and resistance between the gas feed streamline and the substrate. Therefore, increasing Reynolds number and promoting the diffusive transport of free radicals to target substrate will be an efficient way to overcome the limiting step in iCVD process and achieve high deposition rate. Carrier gas N2 was used in iCVD process to obtain high deposition rate. Gas velocity vz as well as Reynolds number can be increased after the introduction of carrier gas. For example, when 10 sccm N2 was introduced into the base-case condition, vz increased from 2.1 ×10-3 m/s to 4.3 ×10-3 m/s, Re doubled from 0.7 to 1.56 and corresponding deposition rate increased from 325 to 725 nm/hr. This 98 increase in deposition rate could also be attributed to low concentration of primary free radicals which decreases the free radical recombination. A hot-zone nozzle was incorporated into our setup to increase deposition rate (Figure 4.16). It is found this modification is very efficient to achieve high deposition rate. Reynolds number increased by more than 20 times with hot-zone nozzle and deposition rate increased from 325 to 1098 nm/hr using the base-case condition. However, the deposition rate increased at the expense of film uniformity. Film thickness measurement showed as-deposited film is only uniform in the center region. Uniformity deviation on a 3-inch-diameter silicon wafer increased to 35% over 28 different positions. a) b) Figure 4.16 A hot-zone nozzle was added to improve deposition rate. a) Original hot-zone design; b) reactor modification with hot-zone nozzle. Introduction of non-reactive solvent-vapor not only results in high molecular weight polymer but also leads to high deposition rate. Figure 4.17 shows how polymer deposition rate depends on flowrate of t-butanol solvent-vapor. Deposition rate increased from 325 to 575 nm/hr after 10 sccm solvent-vapor was introduced into the base-case condition. Deposition rate starts to reach a plateau after solvent-vapor flowrate increased even more. Solvent leads 99 to fast polymerization and higher molecular weight polymers. However, in the deposition with high solvent-vapor flow, the film growth is limited by the concentration of primary free radicals. The condensation or trapping of small solvent molecules into as-deposited polymer films may be another reason to achieve high deposition rate. Deposition rate (nm/hr) 600 Mn:37 Kg/mol 550 Mn:32 Kg/mol 500 450 400 350 0 10 20 30 40 Porogen flow rate (sccm) 50 Figure 4.17 The dependence of deposition rate on solvent flow rate in base-case condition. 4.4 Summary This study demonstrates that poly (methyl methacylate) films can be deposited at relatively low dissociation temperature using an axisymmetric iCVD reactor. Mixtures of monomer, methyl methacrylate and initiator, t-butyl peroxide were fed into a vacuum reactor containing a hot-zone and back-cooled substrate. 100 The resulting as-deposited films have smooth surfaces and exhibit molecular weights ranging from 20-32 kDa. Several reactor design parameters were studied to understand film growth limitations. An Arrhenius relationship (with slope 97 kJ/mol) between the deposition rate and the hot-zone temperature indicates that polymer growth is limited by thermal initiation of free radicals. High deposition rates require good thermal contact between the hot-zone and feed gases. Upon increasing the reactor pressure, the rate of film growth rate exhibits a maximum that is explained using a simple tubular reactor model. At low pressures, free-radical initiation is inefficient in the hot zone, and at high pressures, recombination of free radicals above the substrate limits film growth rate. Higher film growth rates were also observed upon decreasing the substrate temperature. These data suggest that deposition is limited by two effects: monomer adsorption on the substrate and free radical diffusion to the substrate. As expected, increasing the monomer-to-initiator molar feed ratio increased molecular weight. Similar effects were observed by introducing a third, non-reactive component (t-butanol). Several efforts have been made to increase the deposition rate. Introduction of carrier gas increases volumetric flow rate, Reynolds number and deposition rate effectively. Modification to the hot-zone design, i.e. incorporation of hot-zone nozzle increases deposition rate at the expense of film uniformity. Solvent not only leads to longer molecular chain length but also higher deposition rate. Introduction of solvent-vapor also changes the topography of as-deposited films. Under certain circumstances, solvent-vapor promotes the dewetting which will be discussed in detail in the next chapter. 101 Chapter 5. Solvent-Assisted Dewetting During Chemical Vapor Deposition Process 5.1 Introduction 5.1.1 Dewetting and solvent-induced dewetting The film stability and its morphology have been studied extensively for its applications in coatings, adhesives, and dielectric layers and for its fundamental scientific interests. In particular, the dewetting phenomenon of thin polymer films has received much attention. Using spin coating or dip coating, smooth polymer films can be prepared even on nonwettable surface. However, such films are not stable. When they are heated above glass transition temperature (Tg), they will dewet the substrate and are transformed into polymer droplets. It is believed that thick films (greater than microns) may be stable or metastable due to gravity, for thinner films intermolecular forces such as van der Waals forces dominant system and lead to an amplification of fluctuations of the film thickness.132, 133 A classical model of thin film rupture was proposed by Vrij in late 60’s.134 It indicates that thin film rupture by thermal fluctuation is analogous to spindonal decomposition of polymer blends or block copolymer, where height (thickness) fluctuations in this model correspond to composition fluctuations in the polymer blends. However, it has to be mentioned that the surface energy rather than the fluid interfacial energy is the driving force for the film rupture process so this is only a mathematically analog between these two processes. 102 Dewetting is a consequence of how the total excess free energy (G) depends on intermolecular interactions and film thickness h. An expression often used to explain this dependence is: G Aeff 12h 2 S P exp( h ) l [5.1] where Aeff is the effective Hamaker constant, SP is the polar component of the spreading coefficient, and l is a polar decay length.132, 133 Spreading coefficient, which indicates the energy difference between bare and wet substrate, is defined as: S SO ( SL ) [5.2] where γSO, γSL and γ are the solid/air, solid/liquid, and liquid/air interfacial tensions, respectively. If 2 G / h 2 0 then surface fluctuations lower the total excess free energy, and the film spontaneously dewets in accord with spinodal decomposition. The dynamics of surface fluctuation amplification that leads to dewetting has been theoretically worked out by Wyart and Dalliant.135 Figure 5.1 describe the early stage of dewetting, the amplification of height fluctuation in thin films on a solid substrate. 103 Figure 5.1 Schematic drawing of a modulated surface for liquid film on solid substrate. Accordingly, the local film thickness z depends on both position x and time t, and surface undulations grow according to z ( x, t ) h u exp(iqx) exp( Rt ) [5.3] where h is the original film thickness, u is the amplitude of undulations, q is the corresponding wavevector, and R is the fluctuation growth rate. The fastest growing wavevector qM and its growth rate RM scale with film thickness h and liquid viscosity according to: qM 3 / 2 a / h 2 , RM 3a 4 / 4h 5 [5.4] where a 2 Aeff / 6 defines a molecular length scale, is the surface tension. Dewetting occurs if a growing fluctuation reaches the substrate surface (z = 0). Poly(methyl methacrylate) (PMMA) and polystyrene (PS) are the most common system used to study dewetting phenomenon. Experiment was performed 104 by spin-coating a thin layer of PMMA, usually less than 100 nm, onto a PS substrate, annealing it above the glass transition temperature (Tg) of PMMA then observe the morphology change under the microscope.133, 136 Figure 5.2 shows different stages for a typical dewetting process. It usually starts from randomly distributed holes (Figure 5.2 a) then these holes grow in size and touch each other (Figure 5.2 b) to form unstable polygon (Figure 5.2 c). After that, polygon breaks, the materials of polygon are collected into small uniformly distributed droplets (Figure 5.2 d). The final dewetting morphology depends on annealing temperature, time and most importantly, film thickness. (a) (b) (c) (d) Figure 5.2 Different stages for a typical dewetting process.The length of the bar is 100 µm. This figure was reproduced from reference 133. 105 Compared with well-studied thermal-induced dewetting, solvent-induced dewetting has received very little attention. Studies on solutal dewetting have been focused on the experiments where evaporation plays a major role. For example, the polymer solution was spin coated then dewetting occurs as the solvent evaporates.137 Generally speaking, a film can be destabilized by long-range forces, polar interaction and molecular forces in the film having a structure difference.132 However, the main difference between thermal and solvent-driven dewetting is that the cause of instability is the long-range force of van der Waals interactions in the thermal dewetting whereas it is the short-range force of polar interactions in solvent-driven dewetting. In fact, the long-range force in the solvent-driven dewetting becomes a stabilizing rather than a destabilizing factor due to the present of solvent. The presence of these two opposing forces, which enables one to manipulate these forces, is a distinct advantage over the thermal dewetting in the investigation of dewetting behavior. In the solvent-driven dewetting process, the spreading coefficient S could be changed by an order of magnitude with a simple choice of solvent due to the polar interaction. This could be regarded as another advantage of solvent-driven dewetting over the thermal dewetting. To study solvent-induced dewetting, most of the works reported so far use spin coating or dip coating for making polymer films.132, 138, 139 In this study, a different approach, initiated chemical vapor deposition was performed in the presence of solvent-vapor and to understand how solvent-vapor influences polymer film surface topography. As mentioned in the last chapter, solvent-vapor promotes the formation of higher molecular weight polymer, and increases the deposition rate. 106 Depositions performed without solvent-vapor always lead to smooth and featureless films. Introduction of solvent-vapor (t-butanol) during iCVD results in non-equilibrium, thin-film structures reminiscent of dewetted polymer films. In the present study, deposition conditions including solvent-vapor and carrier gas flowrate are systematically adjusted to relate process conditions to the surface topography and droplet size. Observed droplet features arise from a thin-film instability that occurs during early stages of film growth. An autophobic dewetting mechanism is then proposed to describe the topographic development during deposition. This study develops iCVD as a technique to fabricate structured polymer films with tunable surface finish. The ability to control surface morphology of polymer films, in particular poly(methyl methacrylate) may be useful in general as a surface modification tool, and specifically within the semiconductor industry118, 119, for use as biosensors140 and to engineer hydrophobic surfaces141. 5.2 Experimental 5.2.1 Materials Methyl methacrylate (MMA) (Alpha Aesar, 99% purity), t-butyl peroxide (Sigma Aldrich, 97% purity), t-butanol (J. T. Baker, 99.99 % purity) were all used as-received. Three-inch silicon wafers or glass slides were used as deposition substrates. Prior to deposition, wafers (Silicon International) were soaked into a liquid mixture containing 7-parts H2SO4 and 3-parts H2O2 for one hour at 120°C, followed by a 3-minute air-plasma treatment. Alkyl groups were grafted onto 107 cleaned silicon wafers according to a frequently employed procedure.142 Glass slides were cleaned by soaking in a base-bath and then were vacuum-dried before each deposition. 5.2.2 Initiated chemical vapor deposition of PMMA PMMA films were deposited using an axisymmetric vacuum deposition chamber that is described in last chapter. During a typical experiment, the system was first pumped down to a base pressure of 7 Torr, and the hot-zone was heated to the required temperature at 200°C. Then the substrate was cooled and kept at a fixed temperature (-15°C). After the system was maintained at thermal equilibrium for half an hour, monomer, initiator, solvent and nitrogen carrier gasses were fed at set flowrates. In all experiments, the monomer and initiator flowrates were fixed at 4 sccm and 2 sccm, respectively. Solvent and carrier gas flowrates were adjusted between 0 to 20 sccm to study the effects of these two deposition conditions on film topography. In this paper, S and N will be used to describe the flowrates of solvent and nitrogen carrier gas, respectively. For example, S5-N5 indicates the deposition with 5 sccm solvent and 5 sccm N2. After the deposition was complete, the chiller and mass flow controllers were turned off. The reactor chamber was pumped down for an additional half-hour to remove unreacted monomers. 5.2.3 Characterization of iCVD films White light interferometry (Zygo NewView 100) was used to observe the film surface topography and to measure film thickness and deposition rates. The thickness was averaged from measurements of three different substrate positions. 108 Film thickness results from interferometry were confirmed by an optical film measurement device (Filmmetrics, model: F20). To examine the frequency dependence of the feature length scale in the dewetted structure, 1-D power spectral density (PSD) were calculated from the raw interferometry data. This was done by squaring the Fourier transform coefficients of each of the 640 data rows (480 pixels long). All resulting 1-D power spectra were then averaged to determine a representative power spectrum. The averaged PSD is calculated by squaring the averaged power spectra then divided by total length L.143 It is plotted as a function of frequency for the 352 × 264 µm patch. For each set of data, power spectra density calculations were also performed on each of 480 data columns (640 pixels long) and they were in rough agreement. Contact angle measurements were conducted using a VCA Optima XE system. The surface tension of cleaned silicon wafer and silicon wafer grafted with alkyl group were calculated using the Harmonic methods with water, xylene and formamide. Tapping mode AFM was performed with a Digital Instruments (DI) Nanoscope IIIa scanning probe microscope. Topographic (height) and phase contrast images were recorded simultaneously at ambient conditions. Commercial silicon cantilever probe was oscillated at its fundamental resonance frequencies, which ranged between 60 and 90 kHz. The level of tapping force used during imaging is related to the set-point ratio, which is defined as the set-point amplitude to the free-oscillation amplitude. The force level corresponding to set-point ratios of 0.65 (moderate tapping) was investigated in this study. 109 5.3 Results and discussion 5.3.1 Dependence of film morphology on deposition conditions Figure 5.3 shows interference microscope images of polymer films grown without and with a solvent-vapor feed. As-deposited films fabricated without solvent-vapor show relatively smooth and featureless surfaces (Figure 5.3 a). Resulting films have thickness deviation only 3% over 20 measurements on a 3-inch silicon wafer substrate, consistent with our study in the last chapter. The average root-mean-square (RMS) roughness of the control experiment (without solvent) is about 20 nm which is less than one percent of the film thickness. It is well know that PMMA wets SiOx/Si substrates. Deposited PMMA films are thermodynamically stable due to the interfacial energy difference between SiOx/Si 144 (surface free energy: 72 × 10−3 J m−2) and PMMA145 (surface free energy: 32.0 × 10−3 J m−2). However, the introduction of a tert-butanol solvent-vapor feed during the iCVD process significantly influences polymer film topography. T-butanol was selected because it is stable at the hot-zone dissociation temperature (200 °C), and, more importantly, t-butanol solubilizes methyl methacrylate monomer and oligomers, but is insoluble with the corresponding polymer (PMMA). This feature has been leveraged to fabricate asymmetric porous membranes using thermal-induced phase separation (TIPS).120, 121 The resulting morphology of a typical film fabricated using a solvent-vapor flowrate of 10 sccm and a nitrogen (carrier gas) flowrate of 5 sccm (denoted S10-N5) is shown in Figure 5.3 b. Polymer droplet-like features are randomly distributed with average domain size around 20 µm. The spacing between 110 a) 200 -3 100 x10 30 25 20 15 10 5 0 m 0 0 100 200 300 m b) 0.20 0.15 200 0.10 0.05 100 0.00 m 0 0 100 200 300 m c) 0.20 0.15 200 0.10 0.05 100 0.00 m 0 0 100 200 300 m Figure 5.3 Interferometry images of as-deposited PMMA films: (a) deposition without solvent; (b) deposition with t-butanol solvent (S10-N5); and (c) the same film, following annealing at 120 °C for 2 hours. 111 individual droplets is the in range of 20 to 40 µm. The height of these features (z-scale) is about 200 nm which is two orders of magnitude larger than the resolution of the instrument. Remarkably, there is little or no film observed between droplets, i.e. droplets appear to sit directly on the SiOx/Si substrate. The film shown in Figure 5.3 b was thermally annealed above the glass transition of PMMA film to evaluate the stability of the formed features. Following annealing at 120 °C for 2 hours, the features became more spherical as shown in Figure 5.3c. This indicates that during vapor-deposition non-equilibrium features are formed. During annealing, softening of the polymer phase permits the surface tension to act on the droplet-like features. Resulting features are similar to thermal-induced dewetted structures that have been widely studied over the past few decades.133, 136, 146, 147 However, considering the tendency of PMMA to wet silicon dioxide, it is unlikely that the annealed structures are in equilibrium. The spreading of PMMA on substrate is a slow process—on a time-scale much longer than annealing time.139 Qualitatively, observed topologies in Figure 5.3 b may be explained by thin film dewetting that occurs during film growth. Thin film dewetting is of great interest and has been studied experimentally133, 149-153 136, 147, 148 and theoretically135, . Dewetting results when the intermolecular force field is such that the disjoining pressure exceeds the Laplace pressure arising from surface undulations. Dewetting can also occur through other mechanisms including surface nucleation154, 155 , density variation156, and residual stresses157. Autophobic dewetting is dewetting of a fluid on top of a film made of identical molecules. 112 Autophobic dewetting of PMMA films from SiOx / Si can be induced by exposure to a good solvent132, 139. As mentioned in the introduction section, thickness fluctuation of the thin film can be affected by the original film thickness, molecular length scale and surface tension. However, the growth rate of surface instabilities may also be suppressed by the deposition / condensation of new material.158 If the film grows fast enough there may not be enough time for surface instabilities to grow and reach the surface. From the scaling of qM and RM in Equation 5.4, one expects that faster deposition rates and higher film viscosities should suppress thin film dewetting. In the iCVD process, the deposition rate and the thin film viscosity can be adjusted by changing carrier gas and the solvent-vapor flowrates. Figure 5.4 shows how the average mass deposition rate depends on solvent-vapor and nitrogen flowrates. The average mass deposition rate was calculated from interferometry raw data by averaging the film height of all points and multiplying the result by the density of PMMA (1.16 g/cm3).159 This enables the average mass of both wetted and dewetted film to be measured. 113 2 10 -5 Mass Deposition Rate (10 g / cm s) 12 8 : no solvent : 5 sccm solvent : 10 sccm solvent 6 4 2 0 0 5 10 15 20 N2 Flow Rate (sccm) Figure 5.4 Dependence of average mass deposition rate on solvent and carrier gas flow rate. The introduction of carrier gas independently increases deposition rate. Solvent-vapor flow increases deposition rate, and lowers viscosity at the surface of the growing film. The use of nitrogen carrier gas increased the deposition rate regardless of the presence of solvent-vapor. The carrier gas increases the volumetric flow rate and enables inertial forces to transport active free radicals closer to the substrate, reducing diffusive resistance. A similar effect was mentioned in the last chapter where the reactor base-pressure was shown to influence the gas residence time and velocity through the hot-zone. The observed increase in deposition rate may also be attributed to lower concentration of primary free radicals, due to dilution with the carrier gas, which decreases the recombination rate. Solvent-vapor also increases the average mass deposition rate, and this increase is primarily attributed to reduced mass transport resistance at the substrate. If the total mass deposition rate is plotted against total (solvent + 114 carrier gas) flowrate, then the data in Figure 5.4 nearly fall onto a single line suggesting the mechanism of accelerated film growth is the same. However, the solvent also interacts with the substrate. Assuming the solvent-vapor obeys Henry’s law, then the partial pressure of solvent-vapor establishes equilibrium with a surface liquid layer on the substrate. The presence of solvent within the film lowers the film’s viscosity and increases the polymer chain mobility. While the amount of solvent within the growing film can not be quantified, the presence of solvent has been shown to lead to faster polymerization and higher molecular weight polymers. If the features in Figure 5.3 originate from thin-film dewetting, then, according to Equation 5.4, both the deposition rate and surface viscosity should affect the length-scale of surface fluctuations that lead to dewetting. The carrier gas flowrate can be used to independently vary the film deposition rate, however solvent-vapor increases film growth rate while lowering surface viscosity. Figure 5.5 indicates how film topography depends on solvent-vapor and nitrogen flowrates. All images are 352 264 m and all z-scale are the same and abide by the color legend in the upper right. All films grown without solvent-vapor are relatively smooth with measured surface roughness less than 20 nm. Films grown using solvent-vapor feed and a low rate of carrier gas supply (<10 sccm N2) show evidence of dewetting. The length scale of resulting features is greater for films deposited with higher solvent flowrate. However, as the carrier gas flowrate increases (> 10 sccm) as-deposited films tend to be smooth again. These overall trends are qualitatively consistent with Equation 5.4 and the premise that high deposition rates and higher viscosity may suppress dewetting. 115 Figure 5.5 As-deposited film morphology at different solvent-vapor and carrier gas flowrates. 116 Smaller surface fluctuations which are not clearly visible in Figures 5.3 and 5.5 were observed in some of the stable films (S10-N10 and S5-N10), though the amplitudes (z-scale) of these features are less than 10% of those in dewetted films. It is not clear what causes these feature, though they may be due to surface fluctuations at early times of film growth that were dampened by continued deposition. 1D Power spectra of vapor deposited films were calculated to quantify the observed roughness and to determine the spatial periodicity of dewetted features.160 Resulting power spectral density (PSD) curves are plotted against the surface frequency f in Figure 5.6. Dewetted films have PSDs that exhibit a plateau at low frequencies and power-law decay at higher frequencies. The measured PSD were fit to the k-correlation model: PSD A (1 B f 2 ) ( C 1) / 2 2 where A, B, and C are function parameters. [5.5] Fitted parameter values are summarized in Table 5.1. The fitted value of A is related to the low frequency power (the plateau) and the peak-to-valley heights of dewetted structures. The parameter B (m) is related to the transition from the plateau region to the downward-sloping linear region. The frequency of this “knee” indicates the average in-plane correlation length between PMMA features. The parameter C corresponds to the inverse slope at high frequencies and is related to the growth mechanism of the thin film. 117 Figure 5.6 shows the deposited films’ power spectral density dependence on carrier gas and solvent-vapor flows. The smoothest film prepared without any solvent, S0-N0, has no plateau, and its PSD decays nearly linearly- indicative of uncorrelated fractal growth. Dewetted films, on the other hand, all exhibit a clear plateau level (A) greater than 0.1 µm3. For these films, the plateau level generally decreases with carrier gas and increases with solvent flow. The wetted film, S5-N20, exhibits a plateau even though interference microscopy revealed a continuous film. This sample exhibits a correlation length (B) similar to that obtained from dewetted films S5-N0 and S5-N5. While S5-N20 did not ultimately dewet, the same surface fluctuations may be present that drive dewetting. S5-N10 appears to have a similar crossover point however PSD fitting was not successful due to a topographic defect in the image. 118 a) 2 10 S5-N0 1 3 Power Spectral Density (m ) 10 0 10 -1 10 -2 S5-N5 10 -3 10 S5-N20 -4 10 -5 10 -6 10 -7 10 S5-N10 -8 10 3 4 5 6 0.01 2 3 4 5 6 0.1 2 3 4 5 6 -1 Spatial Frequency (m ) b) 2 10 S5-N0 1 10 0 3 Power Spectral Density (m ) 10 -1 10 -2 10 -3 10 S10-N0 -4 10 -5 10 -6 10 -7 10 S0-N0 -8 10 -9 10 -10 10 3 4 5 6 0.01 2 3 4 5 6 0.1 2 3 4 5 6 -1 Spatial Frequency (m ) Figure 5.6 Power spectra density of films deposited with: a) different carrier gas (N2) and b) different solvent-vapor flowrates. Dashed lines are least-squares fits to the k-correlation model. 119 Table 5.1 Parameters from fitting k-correlation model for PSD plot of dewetted and stable films. iCVD Film S5-N0 S5-N5 S10-N0 S5-N20 A (µm3) 22.840 0.405 63.096 7.830×10-5 B (µm) 7.500 7.271 100.995 6.378 C 3.413 1.756 2.500 1.580 Dewetted features on hydrophobic surfaces are also observed when PMMA is deposited in the presence of t-butanol solvent-vapor. For these experiments the silicon substrate was modified by treating with the silane-coupling agent tert-butyl trichlorosilane. The presence of a hydrophobic surface was verified by measuring the water contact angle. Before treatment, the water contact angle was measured to be 11.3 ± 5°, and after treatment, it was measured to be 112.3 ± 5°. Figure 5.7 shows two process diagrams that indicate whether dewetting or wetting was observed—one diagram is for hydrophobically-modified substrates and one is for unmodified SiOx/Si substrates. In constructing the diagrams, dewetted films were easily differentiated from wetted films. Dewetted films all contain features with z-scale (height) in the range of 0.1~0.2 µm, which is about a factor of ten higher than features observed in the smooth, wetted films. In both diagrams, dewetting is observed at low carrier gas flow (lower deposition rates) and higher solvent-vapor contents. However, the dewetted region is larger for films deposited onto the hydrophobic surface. On the hydrophobic surface, experiments focused on trying to locate boundary between the wetting and 120 a) Solvent Flow Rate (sccm) 10 5 0 0 5 10 15 N2 Flow Rate (sccm) 20 0 5 10 15 N2 Flow Rate (sccm) 20 b) Solvent Flow Rate (sccm) 10 8 6 4 2 0 Figure 5.7 Process diagram indicating whether dewetting was observed in solvent-vapor assisted iCVD growth of glassy PMMA films. Depositions were performed on a) plasma-treated silicon wafer substrates and b) hydrophobically modified substrates. The symbols × and ○ indicate dewetted and wetted films respectively. 121 dewetting regions. The correlation length of dewetted features was slightly larger for films grown on the modified surface (data not shown). Water contact angles of vapor deposited films were studied to examine how the textured surfaces described affect wettability. These experiments were only performed on coatings deposited onto SiOx/Si substrates. As a control, the water contact angle on plasma-cleaned SiOx/Si was measured to be 11.6 ± 5°. Vapor-coated PMMA, without solvent, exhibited a water contact angle of 63.3 ± 5° which is somewhat lower than the accepted value of 68°. The dewetted coatings S10-N0 and S5-N5 are more hydrophobic and exhibit higher contact angles of 70.0° and 101.3°, respectively. The water contact angle on S10-N0 is very close to that on smooth PMMA, suggesting either that the vapor coating completely covered the wafer surface, or that the texture of the PMMA features can prevent water from contacting the area between the droplets. If, the latter were true, one would expect a much higher contact angle due to air-water contacts on the underside of the water droplet. This may occur for S5-N5—a film with a finer dewetted structure and a much greater water contact angle. Using Cassie’s law, the fraction of PMMA surface in contact with water can be deduced. From the contact angle data for S5-N5, this fraction is 59% which is in rough agreement with the area of polymer surface observed from interferometry shown in Figure 5.5. These contact angle measurements indicate that simultaneous vapor deposition and dewetting my offer a simple approach to create more hydrophobic surfaces. 122 5.3.2 Proposed Dewetting Mechanism During iCVD the film thickness is continuously growing. The growth rate is limited by transport of initiator to the surface and the reaction kinetics of polymerization.33 This situation is fundamentally different from the majority of polymer dewetting studies. Typically constant-thickness polymer films are thermally annealed or exposed to solvent-vapors, and the entire dewetting process is followed with time-dependent imaging. In situ studies are not feasible with the current iCVD setup. While the proposed mechanism shown in Figure 5.8 offers an explanation for observed topographies, it also draws from several studies of thermal and solutal dewetting on uniformly-thick polymer films. Monomer, initiator and solvent first condense on the cooled substrate to form a physisorbed liquid monolayer. To a rough approximation, the concentration of each component is assumed to be proportional to the ratio Pi/Pi,sat where Pi is the gas-phase pressure and Pi,sat is the component’s saturated vapor pressure.124 Using pure-component vapor pressures at 0 °C the composition of the physisorbed layer can be estimated.126, 161 If this approximation is made on S10-N10, for example, one finds the layer is mostly t-butanol (68%), methyl methacrylate (22%), and the balance is di-tert-butyl-peroxide. This liquid layer is less than 2 nm thick and may interact with the substrate to form a stable layer.151 When PMMA chains begin to form within the thin liquid layer, they adhere to the SiOx / Si substrate.150 A layer of polymer forms and is firmly anchored to the SiOx/Si substrate due to favorable interactions between PMMA and surface hydroxyl groups. As polymerization proceeds, the underlying SiOx/Si substrate becomes saturated with PMMA. More polymer chains form, additional 123 monomer and solvent are deposited, and the liquid layer begins to grow in thickness. At some thickness, the liquid layer becomes unstable and autophobic dewetting occurs from the anchored PMMA layer. Figure 5.8 Proposed dewetting mechanism. Initially a liquid layer (yellow) adsorbs on the surface consisting of predominately methyl methacrylate and t-butanol. As PMMA is polymerized, a strongly anchored layer of PMMA (blue) coats the substrate. The solvent-monomer mixture forms an unstable film on top of the anchored PMMA. (a) If deposition is slow, or if the viscosity is low enough, then surface fluctuations reach the substrate and dewetting occurs, leaving glassy PMMA droplet-like features (green). (b) If deposition is fast enough or the viscosity is high enough, then the time-scale of dewetting is too slow and a uniform coating, which may possess small surface undulations, results. Xue et al. have recently studied solvent-vapor induced dewetting of thin PMMA films on a silicon wafer with a native oxide layer.139 In their study, upon swelling with a good solvent, solvated polymer chains gain mobility and become 124 entropically distinct from tightly-anchored PMMA. This difference in entropy establishes a new interface and drives autophobic dewetting akin to polystyrene thin films dewetting from polystyrene grafted brushes.162, 163 The presence of a tightly-bounded layer is difficult to unambiguously prove, however, there is indirect evidence. The contact angle measurement of water on a dewetted structure (S10-N0) shows a water contact angle of 70° which is much closer to PMMA (63°) than to the bare silicon surface (12°). If bare silicon surface were exposed, the contact angle would be between 12 and 63°. Phase-contrast tapping mode AFM was performed on dewetted PMMA samples and a silicon wafer dip-coated with PMMA, as a control (Figure 5.9). Figure 5.9 a shows the AFM phase-contrast image of as-deposited dewetted PMMA on Si/SiOx substrate. The dewetted polymer droplets show little phase contrast (<5 °) in comparison with the region between them. The phase contrast is determined by many factors including ambient conditions, instrument operating parameters and mostly importantly, the tip-sample interaction. The similarity of phase contrast in the dewetted film indicates that possibility of tightly-bounded PMMA layer between glassy PMMA droplets. In order to evaluate the phase angle difference between PMMA and bare Si/SiOx, Figures 5.9 B and 5.9 b show the height and phase-contrast images of dip-coated PMMA on bare Si/SiOx substrate. The dark and light areas of Figure 5.9 B are presumed to be bare wafer (dewetted regions) and PMMA film. The image shows much higher phase contrast (~50 °) between these two regions. Attempts were also made to use FT-IR microscopy to confirm the presence of a tightly-bound layer. Unfortunately, the instrument did not have sufficient resolution to draw conclusions. 125 Figure 5.9 Topography (left) and phase (right) images for (A, a) dewetted PMMA film on Si/SiOx substrate; (B, b) dip-coated (and partially dewetted) PMMA on bare silicon wafer. Several other dewetting mechanisms may be proposed to explain the observed dewetting during iCVD. Dust particles or compositional variation of the SiOx/Si substrate may lead to microscale wettability contrast and heterogeneous dewetting involving a nucleation growth mechanism.154, 155 Alternatively, density in the thin liquid film may exhibit in-plane spatial variation resulting in van der Waals forces which promote dewetting.156 These mechanisms, however, do not explain how the dewetting length scale depends on solvent and 126 nitrogen flowrate. The rapid formation of residual stress may also induce dewetting during iCVD. As the polymer chains grow, their conformational freedom and free volume change remarkably. Polymers may become trapped in a frozen, non-equilibrium state with significant residual stress stored within the films. These stresses can result in topographic changes, even at temperatures well beneath the glass transition temperature.157 However, the smooth edges of the dewetted structures observed here, and the dependence of the dewetted length-scale on deposition rate, are most consistent with the proposed autophobic dewetting mechanism. Since the solvent-swollen liquid layer is estimated to contain about 60% t-butanol, the presence of solvent in the liquid layer may promote dewetting from the PMMA-anchored layer. The further consider this possibility, contact angle measurements of t-butanol onto SiOx/Si and PMMA-coated substrates were measured to be 9.2 ± 5.0 and 9.7 ± 5.0 ° respectively. The total surface spreading coefficient St containing both dispersive (Sd) and polar (Sp) component is related to the contact angle by: S t S d S P LV (cos 1) [5.6] where LV is the liquid-vapor surface tension of t-butanol (19.6 dynes/cm)164. The effective Hamaker constant can be estimated from: Aeff ( Asubstrate Aliquid )( Aair Aliquid ) [5.7] where Asubstrate, Aliquid, Aair are the pure component Hamaker constants for different phases. Estimates for pure component Hamaker constants165, 166 are: ASiOx = 6.6 10-20 J, APMMA = 7.1 10-20 J, Aair = 0, and the pure Hamaker constant for 127 t-butanol was estimated based on surface tension to be 5.9 10-20 J. The effective Hamaker constants for t-butanol onto SiOx/Si and PMMA are calculated using equation 5.7 to be -0.34 10-20 J and -0.57 10-20 J respectively. By definition Sd has an opposite sign of Aeff, therefore t-butanol’s non-polar interactions promote spreading. Considering this, juxtapose to the measured contact angles, Equation 5.6 suggests that the total spreading coefficient is negative, and polar component of the spreading coefficient from t-butanol destabilizes the thin film. After dewetting, higher molecular weight PMMA forms within the dewetted droplets. These chains are no longer miscible with t-butanol solvent.121 As the polymer phase rejects the solvent it obtains higher viscosity, and eventually a glassy state is formed between the anchored PMMA layer and the solvent-monomer liquid layer. This may explain why solvent-induced dewetting during iCVD results in irregular-shaped (non-equilibrium) polymer features. Also after dewetting, a new surface of anchored PMMA chains is exposed between dewetted droplets. Monomer and solvent deposit on the new surface, chains begin to grow, and the layer thickness grows until it is unstable and dewetting again occurs. This cycle repeats itself over and over—each time additional polymer material is deposited onto the growing polymer droplets. Autophobic dewetting qualitatively explains how film topologies vary with carrier gas and solvent-vapor flowrate. If the film growth rate exceeds the surface instability growth rate then dewetting can be suppressed. In other words, dewetting will not occur if the film growth rate ( h / t ) exceeds the time-derivative of equation 5.3: 128 h Ru exp(i qx) exp( Rt ) t [5.8] However R and qM are sensitive to film thickness and scale with h-5 and h-2, respectively, and therefore one would expect sharp boundaries between wetted and dewetted regions in Figure 5.7. However the boundaries not strictly defined by this inequality (Equation 5.8) because higher solvent-vapor flowrates also reduce the liquid film viscosity and accelerate dewetting. This proposed explanation of dewetting (Figure 5.8) suggests that the lateral length-scale of dewetting, defined by the fasted growing surface fluctuation qM, is independent of time. While time-dependent studies of the dewetting phenomena have not been extensively carried out, there is reason to believe that the lateral length-scale should grow with time. At some point during film growth, the PMMA reaches an insoluble, glassy state. Before this occurs, if the polymer droplets are swollen with solvent and are mobile enough, they may coalesce to form larger droplets. For experiments conducted on the hydrophobically modified surfaces, the surface does not provide hydroxyl groups that can anchor a thin PMMA layer. Therefore, dewetting from hydrophobically modified surfaces may not be driven by short-range polar interactions, but may be due only to long-range apolar interactions, i.e. Aeff > 0. However long-range interactions are difficult to quantify since, neglecting a formed polymer layer, the system contains at least four layers: thin film of monomer and solvent / butyl monolayer / SiOx / Si. Nevertheless, the experimental data indicate dewetting does occur, and, importantly, the 129 length-scale of dewetting is not significantly different from dewetting on SiOx / Si surfaces. “Bump-on-bump” structures shown in Figure 5.10 can also be fabricated and highlight the versatility of solvent-assisted iCVD dewetting. The surface texture was produced by first producing a dewetted film on a clean silicon wafer using conditions (S10-N5) that lead to large periodicity (~30 μm). This surface was characterized and subsequently processed using a different set of conditions (S5-N5) that lead to much smaller periodicity (~5 μm). Both length scales are clearly observed in Figure 5.10. comparing Figure 5.10 to the topography of S10-N5 in Figure 5.5, there appears to be little, if any, late stage ripening of the larger-scale periodicity. White pixels in Figure 5.10 are the smaller dewetted features that formed on top of the coarse features. Interestingly, the smaller features are more frequently observed in the center of the larger features, or situated directly on the substrate between the larger features. If an unstable film forms near a large droplet edge, upon dewetting, the liquid material appears to transport to the larger droplet. The “bump-on-bump” structures are also consistent with the idea of autophobic dewetting. During the second deposition the unstable liquid films dewetted directly from glassy PMMA surfaces. Dewetting from anchored PMMA also occurs in the areas between larger features. 130 0.25 400 200 0.20 0.15 µm 0.10 200 100 0.05 0.00 00 0 200 100 m 400 200 600 300 Figure 5.10 Topography of a “bump-on-bump” structure measured using interferometry. 5.4 Summary This study demonstrates that the iCVD technique can be used to fabricate polymer films with periodic lateral length-scales. When PMMA films are grown on SiOx/Si substrates in the presence of a non-reactive solvent-vapor (t-butanol), droplet-like glassy PMMA features form. Our experiments show that the feature length-scale can be tuned between 5 and 100 µm by adjusting two process parameters: the nitrogen carrier gas flowrate and the solvent-vapor flowrate. The carrier gas affects the overall mass flux at the substrate (deposition rate) and the solvent-vapor also increases the deposition rate, but, more importantly, lowers the viscosity of the liquid surface layer. The observation of droplet-like structures is consistent with an autophobic dewetting of a solvent-monomer mixture, likely containing lower molecular weight PMMA, from a tightly anchored PMMA layer. 131 Dewetting of as-deposited films from hydrophobically modified SiOx/Si surfaces was also observed, indicating that structures can be grown on hydrophobic surfaces as well. In this case, dewetting is attributed to long range dispersion forces between the film and the butyl/SiOx/Si layered substrate. “Bump-on-bump” structures were fabricated through two successive depositions and illustrate how dewetting can be used to create hierarchal surfaces. Dewetting in solvent-assisted, vapor-deposited polymer films represents a simple approach to yielding textured surfaces on a variety of substrates, and may facilitate the development of microporous membranes, super-hydrophobic surfaces, and microfluidic devices. 132 Chapter 6. Summary and Future Work 6.1 Summary Fabrication of structured polymer thin films using vapor deposition polymerization (VDP) was explored. VDP technique is a versatile chemical process to grow macromolecular species directly from gas-phase feeds. It differs from traditional solution-based techniques in many desirable ways because the reactants used are gases. Depending on the polymerization reaction mechanism, VDP can usually be summarized into four categories. Among them, step-growth VDP and chain-growth VDP have attracted a great deal of attention and have been used to deposit polymer films in this thesis. Precise control of morphology and microstructure during thin film deposition is important to produce the desired film quality and is the primary focus of current study. Step-growth VDP, chain-growth VDP and other forms of VDP typically results in smooth, homogeneous films. However, the introduction of non-reactive, immiscible species into vapor deposition process has been proved effective to leverage phase separation and introduce heterogeneity. Solventless VDP approach was first used to fabricate micro-thick polybenzoxazole films in Chapter 2. Co-deposition of pyromellitic dianhydride and 3, 3’-dihydroxybenzidine results in poly (amic acid) precursor, evaporation temperature for each component has to be carefully chosen to achieve stiochiometric balance and high quality films. Curing of as-deposited poly (amic acid) under argon purge leads to the formation of polyimides at about 200 °C and 133 subsequently to partially aromatic polybenzoxazoles at 550 °C. The existence of unreacted monomer in as-deposited poly (amic acid) films and the formation of polyimide and PBO were confirmed by FT-IR studies. TGA studies showed the mass losses due to imidization and decarboxylation, as well as a continuous mass loss which could be attributed to unreacted monomer. Nanoindentation studies showed as-deposited and cured VDP films surprisingly exhibit higher or comparable Young’s modulus and hardness compared with solution-cast films. After the thermal conversion from poly (amic acid) to PBO, modulus and hardness can be improved by a factor of three. The successful fabrication of PBO provides a platform to study simultaneous film growth and phase separation in vapor-deposited condensation polymers including polyimides. Non-reactive, immiscible copper phthalocyanine was then used to grow novel polyimide/CuPc composite films and to change the morphology of as-deposited films in Chapter 3. CuPc particles were evaporated with biphenyl tetracarboxylic dianhydride (BPDA) and oxydianiline (ODA) polyimide precursor monomers. FT-IR and UV/Vis of resulting films showed CuPc particles were embedded in the polyimide matrix. Fine lateral structures at the surface and in the bulk with a length scale of about 100 nm were observed using AFM and TEM, respectively. Embedded CuPc particles initially appear amorphous and undergo partial crystallization upon thermal treatment from poly (amic acid) to polyimide at 200 °C. Crystallized CuPc particles display rod-like morphology which is the characteristic of α-phase CuPc crystallites. The morphology can be adjusted by changing CuPc volume fraction and curing temperature. Stable morphology and high level of interfacial surface area observed in composite films may be useful in 134 developing these n-type polyimides as materials for photovoltaic or OLED devices. Different vapor deposition approach, initiated chemical vapor deposition (iCVD) uses chain-growth mechanism to grow poly (methyl methacrylate) films in Chapter 4. This provides a platform to investigate simultaneous film growth and phase separation in addition polymer films. An axisymmetric, multi-component iCVD reactor was designed and used. Mixture of monomer, methyl methacrylate and initiator, t-butyl peroxide were fed into this reactor containing a hot-zone and back-cooled substrate. As-deposited films show smooth, featureless surfaces and have molecular weight ranging from 20-32 kDa. Several reactor design parameters including hot-zone temperature, reactor base pressure, substrate temperature and monomer/initiator feed ratio were systematically verified to understand film growth limitations. The results from these studies suggest that deposition in this axisymmetric reactor is limited by thermal initiation of free radicals, monomer adsorption on the substrate and diffusion transport of primary free radicals to substrate. Deposition rate increases dramatically when deposition conditions were adjusted to overcome these limitations. The molecular weight of as-deposited polymer can be increased by changing the monomer-to-initiator molar feed ratio or introducing a third, non-reactive component (t-butanol). Introduction of non-reactive, immiscible t-butanol solvent-vapor into deposition process also changes the morphology of as-deposited PMMA films. Under certain circumstances, solvent-vapor promotes dewetting and randomly distributed polymer droplets were observed. In Chapter 5, we demonstrate that the 135 iCVD technique is capable of changing periodic length-scale by more than 20 times by adjusting two process parameters: the nitrogen carrier gas flowrate and the solvent-vapor flowrate. The carrier gas increases the overall mass flux towards the substrate, and solvent-vapor increases the deposition rate as well as lowers the viscosity of the liquid surface layer. The morphology of polymer droplet is consistent with an autophobic dewetting of a solvent-monomer mixture from a tightly anchored PMMA layer. Hierarchal, “bump-on-bump” structure can be fabricated through two successive depositions. Most importantly, solvent-assisted dewetting in vapor-deposited polymer films illustrates a simple way to fabricate textured surface on a variety of substrates, and may be used to develop microporous membranes, super-hydrophobic surfaces, and microfluidic devices. 6.2 Future work As mentioned in Chapter 4, our hot-zone iCVD reactor has several advantages over traditional hot-filament design, but it is far from perfect. Not all the fittings and tubing are heated, therefore, liquid condensation could occur between monomer, initiator sources and reactor chamber due to the temperature difference. This will decrease the deposition efficiency dramatically. Packed hot-zone has bigger thermal mass and its temperature can be accurately controlled which eliminates the hot spots. However, big thermal mass leads to high thermal radiation which increases the substrate temperature undesirably. High substrate temperature can result in desorption of monomer and free radical species, low sticking coefficient and low deposition rate. In addition, vacuum grease was used 136 in the deposition process. This improves the thermal contact between silicon wafer substrate and back-cooled stage, but may contaminate the vapor-deposited PMMA films. Several modifications are necessary to solve these problems. Heated aluminum block has been custom-designed to provide uniform heating not only to the monomer and initiator sources, but also to the fittings and tubings between sources and reactor chamber. Instead of messy vacuum grease, small metal clip will be used to secure silicon wafer substrate and improve the thermal contact. We are also trying to build vapor deposition system without vacuum. Carrier gas containing monomer and initiator flows through a heated nozzle where initiator is thermally dissociated. The nozzle is positioned closely and pointed directly to the substrate. Then the free-radical polymerization occurs on the cooled substrate. This design will simplify the reactor, minimize the thermal radiation and increase the deposition rate. The ultimate goal of our study is to fabricate the porous polymer films combining initiated chemical vapor deposition and polymerization-induced phase separation techniques. However, based on our study, it seems vapor-deposited PMMA is not an appropriate candidate to achieve this goal. Pores are reported to be in the micron range if polymerization-induced phase separation is used to make porous materials. This length scale is comparable to the film thickness we deposited in our study. However, high deposition rate is difficult to achieve in iCVD of PMMA films. Highest deposition rates using hot-zone reactor and hot-filament reactor are both around 1 µm/hr. This relatively low deposition rate is due to the high vapor pressure of MMA monomer and molecules tend to desorb from the substrate. Low deposition rate could also be attributed to the rate of 137 propagation of radical polymerization of MMA which does not favor the production of long PMMA chains.29 Low-vapor-pressure methacrylates, such as glycidyl methacrylate (GMA) or 2-hydroxyethyl methycrylate (HEMA) might be good candidates for making porous films using iCVD technique. Careful consideration of monomer evaporation temperature, type of initiator and porogen species, and substrate temperature may enable porous polymer films to be successfully deposited. The introduction of a forth component, a difunctional monomer as crosslinker, is our next step to help arrest morphologies. The presence of crosslinker minimized the gel particle formation where PIPS is present.167 Likewise, the use of crosslinker in the vapor processing of thin film is expected to facilitate the formation of various morphologies during the coarsening process. Chan and Gleason have recently demonstrated that crosslinked thin films of poly (2-hydroxyethyl methacrylate) could be formed using iCVD technique.28 The resulting hydrogels were prepared to different crosslink densities by adjusting the flow rate of crosslinker and they exhibited swelling behavior that reflects the cross-link density. In a similar fashion, crosslinkers will be utilized to arrest morphologies more rapidly, resulting in structures with smaller, more uniform, periodic length scale. 138 References 1. Martin, T. P.; Kooi, S. E.; Chang, S. H.; Sedransk, K. L.; Gleason, K. K., Initiated Chemical Vapor Deposition of Antimicrobial Polymer Coatings. Biomaterials 2007, 28, (6), 909-915. 2. Hori, S.; Yoshimura, M.; Somiya, S.; Kurita, R.; Kaji, H., Mechanical Properties of ZrO2-toughen Al2O3 Ceramics from CVD Powders. Journal of Materials Science Letters 1985, 4, (4), 413-416. 3. Li, Y.; Kinloch, L.; Windle, A. H., Direct Spinning of Carbon Nanotube Fibers from Chemical Vapor Deposition Synthesis. Science 2004, 304, (5668), 276-278. 4. Goela, J. S.; Taylor, R. L., Monolithic Materials Fabrication by Chemical Vapor Deposition. Journal of Materials Science 1998, 23, (12), 4331-4339. 5. Zaharias, G. A.; Shi, H. H.; Bent, S. F., Characterization of Polyconjugated Thin Tilms Synthesized by Hot-wire Chemical Vapor Deposition of Aniline. Thin Solid Films 2006, 501, (1-2), 341-345. 6. Pyo, S. W.; Lee, D. H.; Koo, J. R.; Kim, J. H.; Shim, J. H.; Kim, J. S.; Kim, Y. K., Electrical Effect in Organic Thin-Film Transistors Using Polymerized Gate Insulators by Vapor Deposition Polymerization (VDP). Synthetic Metals 2005, 154, (1-3), 141-144. 7. Strijkova, V.; Dimov, D.; Paskaleva, A.; Zhivkov, I.; Spassova, E.; Assa, J.; Danev, G., Electrical Properties of a Thin Layer Polyimide Matrix. Journal of Optoelectronics and Advanced Materials 2005, 7, (3), 1319-1322. 139 8. Green, Z. I.; Chen, X.; Papastrat, A.; Zou, L.; Anthamatten, M., Morphology of Vapor Deposited Polyimides Containing Copper Phthalocyanine. Chemical Vapor Deposition 2009, in press. 9. Roualdes, S.; Durand, J., Experimental Design and Modelling in the Investigation of PECVD Parameters Effects on the Structural and Gas Transport Properties of Plasma Polysiloxane Membranes. Journal of Membrane Science 2004, 230, (1-2), 39-48. 10. Im, S. G.; Kim, B. S.; Lee, L. H.; Tenhaeff, W. E.; Hammond, P. T.; Gleason, K. K., A Directly Patternable, Click-Active Polymer Film via Initiated Chemical Vapor Deposition. Macromolecular Rapid Communications 2008, 29, 1648-1654. 11. Lau, K. K. S.; Gleason, K. K., Particle Surface Design using an All-Dry Encapsulation Method. Advanced Materials 2006, 18, 1972-1977. 12. Dobkin, D. M.; Zuraw, M. K., Principles of Chemical Vapor Deposition. Kluwer Academic Publishers: Norwell, MA, 2003; p 1-5. 13. Anthamatten, M.; Lau, K. K. S., Vapor Deposition Polymerization. Encyclopedia of Chemical Processing ( In Press) 2008. 14. Mittal, K. L.; Ghosh, M. K., Polyimides: Fundamentals and Applications. Marcel Dekker: New York, 1996. 15. Sato, M.; Iijima, M.; Takahashi, Y., Photoresist Characteristics of Polyurea Films Prepared by Vapor Deposition Polymerization. Thin Solid Films 1997, 308-309, 90-93. 16. Anthamatten, M.; Letts, S. A.; Day, K.; Cook, R. C.; Gies, A. P.; Hamilton, T. P.; Nonidez, W. K., A Investigation of Solid-state Amidization and 140 Imidization Reactions in Vapor-Deposited Poly(amic acid). Journal of Polymer Science Part A: Polymer Chemistry 2004, 42, (23), 5999-6010. 17. Iijima, M.; Takahashi, Y., Vapor Deposition Polymerization: A study on Film Formation in Reaction of Pyromellitic Anhydride and Bis(4-aminophenyl) Ether. Macromolecules 1989, 22, 2944-2946. 18. Salem, J. R.; Sequeda, F. O.; Duran, J.; Lee, W. Y.; Yang, R. M., Solventless Polyimide Films by Vapor Deposition. Journal of Vacuum Science & Technology A: Vacuum, Surface, and Films 1986, 4, (3), 369-374. 19. Tsai, F. Y.; Blanton, T. N.; Harding, D. R.; Chen, S. H., Temperature Dependence of the Properties of Vapor-Deposition Polyimides. Journal of Applied Physics 2003, 93, (7), 3760-3764. 20. Chen, X.; Anthamatten, M.; Harding, D. R., Vapor Deposition and Curing of Polybenzoxazole Precursors. Macromolecules 2006, 39, (22), 7561-7565. 21. Ojeda, J. R.; Martin, D. C., High-Resolution Microscopy of PMDA-ODA Polyimide Single Crystals. Macromolecules 1993, 26, (24), 6557-6565. 22. Gies, A. P.; Nonidez, W. K.; Anthamatten, M.; Cook, R. C., A Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry Study of the Imidization of Vapor-Deposited ODA-PMDA Poly(amic acid). Macromolecules 2004, 37, (16), 5923-5929. 23. Loo, L. S.; Gleason, K. K., Hot Filament Chemical Vapor Deposition of Polyoxymethylene as a Sacrificial Layer for Fabricating Air Gaps. Electrochemical and Solid-State Letters 2001, 4, (11), G81-G84. 141 24. Lau, K. K. S.; Caulfield, J. A.; Gleason, K. K., Structure and Morphology of Fluorocarbon Films Grown by Hot Filament Chemical Vapor Deposition. Chemistry of Materials 2000, 12, (10), 3032-3037. 25. Limb, S. J.; Labelle, C. B.; Gleason, K. K.; Edell, D. J.; Gleason, E. F., Growth of Fluorocarbon Polymer Thin Films with High CF2 Fractions and Low Dangling Bond Concentrations by Thermal Chemical Vapor Deposition. Applied Physics Letters 1996, 68, (20), 2810-2812. 26. Lewis, H. G. P.; Caulfield, J. A.; Gleason, K. K., Perfluorooctane Sulfonyl Fluoride as an Initiator in Hot-Filament Chemical Vapor Deposition of Fluorocarbon Thin Films. Langmuir 2001, 17, 7652-7655. 27. Mao, Y.; Gleason, K. K., Hot filament chemical vapor deposition of poly(glycidyl methacrylate) thin films using tert-butyl peroxide as an initiator. Langmuir 2004, 20, (6), 2484-2488. 28. Chan, K.; Gleason, K. K., Initiated Chemical Vapor Deposition of Linear and Cross-linked Poly(2-hydroxyethyl methacrylate) for Use as Thin-Film Hydrogels. Langmuir 2005, 21, (19), 8930-8939. 29. Chan, K.; Gleason, K. K., Initiated CVD of poly(methyl methacrylate) thin films. Chemical Vapor Deposition 2005, 11, (10), 437-443. 30. Ma, M.; Mao, Y.; Gupta, M.; Gleason, K. K.; Rutledge, G. C., Superhydrophobic Fabrics Produced by Electrospinning and Chemical Vapor Deposition. Macromolecules 2005, 38, (23), 9742-9748. 31. Chan, K.; Gleason, K. K., Photoinitiated Chemical Vapor Deposition of Polymeric Thin Films Using a Volatile Photoinitiator. Langmuir 2005, 21, (25), 11773-11779. 142 32. Martin, T. P.; Sedransk, K. L.; Chan, K.; Baxamusa, S. H.; Gleason, K. K., Solventless Surface Photoinitiated Polymerization: Grafting Chemical Vapor Deposition (gCVD). Macromolecules 2007, 40, 4586-4591. 33. Chen, X.; Anthamatten, M., Vapor Deposition Polymerization of Poly(Methyl Methacrylate) in an Axisymmetric Vacuum Reactor. Polymer 2008, 49, 1823-1830. 34. Yamada, B.; Zetterlund, P. B., Handbook of Radical Polymerization. Wiley-Interscience: New York, 2002; p 117-186. 35. Lau, K. K. S.; Gleason, K. K., Initiated Chemical Vapor Deposition (iCVD) of Poly(alkyl acrylates): A Kinetic Model. Macromolecules 2006, 39, (10), 3695-3703. 36. Venkatasubramanian, R.; Siivola, E.; Colpitts, T.; O'Quinn, B., Thin-film Thermoelectric Devices with High Room-temperature Figures of Merit. Nature 2001, 413, 597-602. 37. Hwang, C. S.; Park, S. O.; Cho, H. J.; Kang, C. S.; Kang, H. K.; Lee, S. I.; Lee, M. Y., Deposition of Extremely Thin (Ba, Sr) TiO3 Thin Films for Ultra-large-scale Integrated Dynamic Random Access Memory Application. Applied Physics Letters 1995, 67, 2819-2821. 38. Stolt, L.; Hedstrom, J.; Kessler, J.; Ruckh, M.; Velthaus, K.; Schock, H., ZnO/CdS/CuInSe2 Thin-film Solar Cells with Improved Performance. Applied Physics Letters 1993, 62, 597-599. 39. Fu, Y.; Huang, W.; Du, H.; Huang, X.; Tan, J.; Gao, X., Charaterization of TiNi Shape-memory Alloy Thin Films for MEMS Applications. Surface and Coatings Technology 2001, 145, (1-3), 107-112. 143 40. Kopp, V. I.; Genack, A. Z.; Zhang, Z., Large Coherence Area Thin-Film Photonic Stop-Band Lasers. Physical Review Letter 2001, 86, (9), 1753-1756. 41. D'Andrade, B. W.; Forrest, S. R., White Organic Light-Emitting Devices for Solid-State Lighting. Advanced Materials 2004, 16, (18), 1585-1595. 42. Schock, H. W., Thin Film Photovoltaics. Applied Surface Science 1996, 92, 606-616. 43. Shi, Y.; Liu, J.; Yang, Y., Device Performance and Polymer Morphology in Polymer Light Emitting Diodes: The Control of Thin Film Morpholgy and Device Quantum Efficiency. Journal of Applied Physics 2000, 87, 4254-4263. 44. Ueda, K.; Tabata, H.; Kawai, T., Magnetic and Electric Properties of Transition-Metal-Doped ZnO Films. Applied Physics Letters 2001, 79, (7), 988-990. 45. Cantalini, C.; Sun, H. T.; Faccio, M.; Pelino, M.; Santucci, S.; Lozzi, L.; Passacantando, M., NO2 Sensitivity of WO3 Thin Film Obtained by High Vacuum Thermal Evaporation. Sensors and Actuators B: Chemical 1996, 31, (1-2), 81-87. 46. Takeda, S.; Suzuki, S.; Odaka, H.; Hosono, H., Photocatalytic TiO2 Thin Film Deposited onto Glass by DC Magnetron Sputtering. Thin Solid Films 2001, 392, (2), 338-344. 47. Hu, J.; Gordon, R. G., Textured Aluminum-Doped Zinc Oxide Thin Films from Atmospheric Pressure Chemical Vapor Deposition. Journal of Applied Physics 1992, 71, (2), 880-890. 144 48. Barabasi, A. L.; Stanley, H. E., Fractual Concepts in Surface Growth. In Cambridge University Press: New York, 1995. 49. Karunasiri, R. P. U.; Bruinsma, R.; Rudnick, J., Thin-Film Growth and the Shadow Instability. Physical Review Letter 1989, 62, (7), 788-791. 50. Zhao, Y. P.; Drotar, J. T.; Wang, G. C.; Lu, T. M., Morphology Transition during Low-Pressure Chemical Vapor Deposition. Physical Review Letters 2001, 87, (13), 136102. 51. Zhao, Y. P.; Wang, G. C.; Lu, T. M., Characterization of Amorphous and Crystalline Rough Surface: Principles and Applications. Academic Press: New York, 2001. 52. Lee, C.; Sazonov, A.; Nathan, A., High-Mobility Nanocrystalline Silicon Thin-Film Transistors Fabicated by Plasma-Enhanced Chemical Vapor Deposition. Applied Physics Letters 2005, 86, (22), 222106-222108. 53. Eyal, A. M.; Hajdu, K.; Hazan, B.; Edelstein, D., Chemically Induced Porogen Decomposition in Premembranes for Porogen Derived Membranes. Journal of Applied Polymer Science 1992, 46, 1613-1620. 54. Eyal, A. M.; Hazan, B.; Hajdu, K.; Edelstein, D., Premembranes for Porogen-Derived Membranes and Thermal Decomposition of Porogens. Journal of Applied Polymer Science 1992, 45, 1065-1074. 55. Heilmann, A., Plolymer Films with Embedded Metal Nanoparticles. 1 ed.; Springer: New York, 2002; p 1-7. 56. Elicabe, G. E.; Larrondo, H. A.; Williams, R. J. J., Light Scattering in the Course of a Polymerization-Induced Phase Separation by a Nucleation-Growth Mechanism. Macromolecules 1998, 31, (23), 8173-8182. 145 57. Martina, A. D.; HIlborn, J. G.; Muhlebach, A., Macroporous Cross-Linked Poly(dicyclopentadiene). Macromolecules 2000, 33, (8), 2916-2921. 58. Rogers, M. E.; Long, T. E., Synthetic Methods in Step-Growth Polymers. Wiley-Interscience: New York, 2003. 59. Sroog, C. E., History of the Invention and Development of the Polyimides. In Polyimides Fundamentals and Applications, Ghosh, M. K.; Mittal, K. L., Eds. Marcel Dekker: New York, 1996; pp 1-6. 60. Vogel, H.; Marvel, C. S., Polybenzimidazoles, New Thermallly Stable Polymers. Journal of Polymer Science 1961, 50, 511-539. 61. Moyer, W. W.; Cole, C.; Anyos, T., Aromatic Polybenzoxazoles. Journal of Polymer Science Part A: Genernal papers 1965, 3, 2107-2121. 62. Clair, A. K.; Johnston, N. J., Ether Polyphenylquinoxalines. II. Polymer Synthesis and Properties. Journal of Polymer Science: Polymer Chemistry Edition 1977, 15, 3009-3021. 63. Choe, E. W.; Kim, Mechanical-Properties S. of N., Synthesis, Spinning, and Fiber "Poly(Para-Phenylenebenzobisoxazole). Macromolecules 1981, 14, (4), 920-924. 64. Davies, R. J.; Eichhorn, S. J.; Riekel, C.; Young, R. J., Crystal Lattice Deformation in Single Poly(p-phenylene benzobisoxazole) Fibres. Polymer 2004, 45, (22), 7693-7704. 65. Kitagawa, T.; Ishitobi, M.; Yabuki, K., An Analysis of Deformation Process on Poly-p-phenylenebenzobisoxazole Fiber and a Structural Study of the New High-modulus Type PBOHM Plus Fiber. Journal of Polymer Science Part B-Polymer Physics 2000, 38, (12), 1605-1611. 146 66. So, Y. H.; Heeschen, J. P.; Murlick, C. L., A Mechanistic Study of Polybenzoxazole Formation with Model Compounds. Macromolecules 1995, 28, (21), 7289-7290. 67. Tullos, G. L.; Powers, J. M.; Jeskey, S. J.; Mathias, L. J., Thermal Conversion of Hydroxy-containing Imides to Benzoxazoles: Polymer and Model Compound Study. Macromolecules 1999, 32, (11), 3598-3612. 68. Liu, X.; Yu, W., Evaluating the Thermal Stability of High Performance Fibers by TGA. Journal of Applied Polymer Science 2006, 99, 937-944. 69. Kitagawa, T.; Murase, H.; Yabuki, K., Morphological Study on poly-p-phenylenebenzobisoxazole (PBO) Fiber. Journal of Polymer Science Part B-Polymer Physics 1998, 36, (1), 39-48. 70. Roitman, D. B.; Wessling, R. A.; McAlister, J., Characterization of Poly(p-phenylene-cis-benzobisoxazole) in Methanesulfonic Acid. Macromolecules 1993, 26, 5174-5184. 71. Feng, D. D.; Wang, S. F.; Zhuang, Q. X.; Wu, P. P.; Han, Z. W., Semi-empirical calculation and spectroscopic study of protonated poly(p-phenylene benzobisoxazole) models. Polymer 2004, 45, (26), 8871-8879. 72. Hsu, S. L. C.; Chang, K. C.; Huang, Y. P.; Tsai, S. J., A Novel Synthesis Method for the Preparation of Aromatic Poly(imide benzoxazole) from Trimellitic Anhydride Chloride and bis(o-aminophenol). Journal of Applied Polymer Science 2003, 88, (10), 2388-2391. 147 73. Kim, T. K.; Choi, K. Y.; Lee, K. S.; Park, D. W.; Jin, M. Y., Thermal Conversion of t-butyloxycarbonyloxy Attached Polyamides to Polybenzoxazoles. Polymer Bulletin 2000, 44, (1), 55-62. 74. Chang, J. H.; Park, K. M.; Lee, S. M.; Oh, J. B., Two-step Thermal Conversion from Poly(amic acid) to Polybenzoxazole via Polyimide: Their Thermal and Mechanical Properties. Journal of Polymer Science Part B-Polymer Physics 2000, 38, (19), 2537-2545. 75. Iijima, M.; Takahashi, Y.; Inagawa, K.; Itoh, A., In Vacuo Synthesis of Aromatic Polyamides. J. Vac. Sci. Jpn. 1985, 28, (5), 437-439. 76. Jandke, M.; Kreger, K.; Strohriegl, P., Poly(phenylquinoxalines) by Vapor Deposition Polymerization. Synthetic Metals 2000, 111-112, 221-223. 77. Marata, H., Multi-layered Polymer Light-Emitting Devices Prepared by Vapor Deposition Polymerization. Synthetic Metals 2001, 121, 1679-1680. 78. Roberts, C. C.; Letts, S. A.; Saculla, M. D.; Hsieh, E. J.; Cook, R. C., Polyimide Films from Vapor Deposition: Toward High Strength, NIF Capsules. Fusion Technology 1999, 35, 138-146. 79. Tsai, F. Y.; Harding, D. R.; Chen, S. H.; Alfonso, E. L., Effects of Processing Conditions on the Quality and Properties of Vapor-Deposited Polyimide Shells. Fusion Technology 2002, 41, 178. 80. Staff, P. D. L., Effect of Creep and Other Time Related Factors on Plastics and Elastomers. William Andrew Publishing/Plastics Design Library: 1991. 81. Sharda, J.; Deenaddayalu, C.; Mobasher, B.; Rajan, S. D., Modeling of Multilayer Composite Fabrics for Gas Turbine Engine Containment Systems. Journal of Aerospace Engineering 2006, 19, (1), 38-45. 148 82. Knight, A. K.; Tsai, F.-Y.; Bonino, M. J.; Harding, D. R., Suitability of Different Polyimide Capsule Materials for Use as ICF Targets. Fusion Science & Technology 2004, 45, 188. 83. Schab-Balcerzak, E.; Jikei, M.; Kakimoto, M., Thermal Rearrangement of Poly(o-hydroxyimide)s Synthesized from 4,6-Diaminoresorcinol Dihydrochloride. Polymer Journal 2003, 35, (2), 208-212. 84. Bulletin GS-96-7 General Specifications: Kapton Polyimide Film. In DuPont High Performance Materials: Circleville, OH. 85. Chang, J. H.; Chen, M. J.; Farris, R. J., Effect of Heat Treatment on the Thermal and Mechanical Properties of a Precursor Polymer: Polyhydroxyamide. Polymer 1998, 39, (23), 5649-5654. 86. Dingemans, T. J.; Picken, S. J.; Murthy, N. S.; Mark, P.; StClair, T. L.; Samulski, E. T., Wholly Aromatic Ether-Imides. Potential Materials for n-Type Semiconductors. Chemistry of Materials 2004, 16, (6), 966-974. 87. Singh, T. B.; Erten, S.; Gunes, S.; Zafer, C.; Turkmen, G.; Kuban, B.; Teoman, Y.; Sariciftci, N. S.; Icli, S., Soluble Derivatives of Perylene and Naphthalene Diimide for n-Channel Organic Field-Effect Transistors. Organic Electronics 2006, 7, (6), 480-489. 88. Kato, Y.; Iba, S.; Teramoto, R.; Sekitani, T.; Someya, T.; Kawaguchi, H.; Sakurai, T., High Mobility of Pentacene Field-Effect Transistors With Polyimide Gate Dielectric Layers. Applied Physics Letters 2004, 84, (19), 3789-3791. 149 89. Kajii, H.; Taneda, T.; Ohmori, Y., Organic Light-Emitting Diode Fabricated on a Polymer Substrate for Optical Links. Thin Solid Films 2003, 438-439, 334-338. 90. Lozano, A. E.; Abajo, J.; Campa, J. G.; Guillen, C.; Herrero, J.; Gutierrez, M. T., Thin-Film Polyimide/Indium Tin Oxide Composites for Photovoltaic Application. Journal of Applied Polymer Science 2006, 103, (6), 3491-3497. 91. Tsai, F. Y.; Harding, D. R.; Chen, S. H.; Blanton, T. N., High-Permeability Fluorinated Polyimide Microcapsules by Vapor Deposition Polymerization. Polymer 2003, 44, (4), 995-1001. 92. Zou, L.; Anthamatten, M., Synthesis and Characterization of Polyimide-Polysiloxane Segmented Copolymers for Fuel Cell Applications. Journal of Polymer Science Part A: Polymer Chemistry 2007, 45, (16), 3747-3758. 93. Kubono, A.; Higuchi, H.; Umemoto, S.; Okui, N., Direct Formation of Polyimide Thin Films by Vapor Deposition Polymerization. Thin Solid Films 1993, 232, (2), 256-260. 94. Law, K. Y., Organic Photoconductive Materials: Recent Trends and Developments. Chem. Rev. 1993, 93, (1), 449-486. 95. Eley, D. D., Phthalocyanines as Semiconductors. Nature 1948, 162, 819-819. 96. Grubbs, R. B., Hybrid Metal-Polymer Composites from Functional Block Copolymers. Journal of Polymer Science Part a-Polymer Chemistry 2005, 43, 4323-4336. 150 97. Bao, Z.; Lovinger, A. J.; Dodabalapur, A., Organic Field-Effect Transistors with High Mobility Based on Copper Phthalocyanine. Applied Physics Letters 1996, 69, (20), 3066-3068. 98. Bundgaard, E.; Krebs, F. C., Low Band Gap Polymers for Organic Photovoltaics. Solar Energy Materials ans Solar Cells 2007, 91, (11), 954-985. 99. Nelson, J., Organic Photovoltaic Films. Current Opinion in Solid State and Materials Science 2002, 6, 87-95. 100. Nguyen, L. H.; Hoppe, H.; Erb, T.; Gunes, S.; Gobsch, G.; Sariciftci, N. S., Effects of Annealing on the Nanomorphology and Performance of Poly(alkylthiophene): Fullerene Bulk-Heterojunction Solar Cells. Adv Funct Mater 2007, 17, (7), 1071-1078. 101. Iwase, T.; Haga, Y., Photovoltaic Characteristics of TCNQ-Incorporated CuPc-Poly(p-phenylene) Composite Films. Journal of Materials Science: Materials In Electronics 2004, 15, 617-621. 102. Dimov, D.; Strijkova, V.; Karamancheva, I.; Zhivkov, I.; Tsenov, I.; Spassova, E.; Danev, G., Copper Phthalocyanine as Quest in a Thin Layer Polyimide Matrix. Journal of Optoelectronics and Advanced Materials 2005, 7, (3), 1445-1449. 103. Kim, J. E. S.; Lim, E.; Lee, K.; Cha, D.; Friedman, B., Effects of Substrate Temperature on Copper(II) Phthalocyanine Thin Films. Applied Surface Science 2003, 205, (1-4), 274-279. 151 104. Karan, S.; Malik, B., Effects of Annealing on the Morphology and Optical Property of Copper(II) Phthalocyanine Nanostructured Thin Films. Solid State Communications 2007, 43, (6-7), 289-294. 105. Achar, B. N.; Lokesh, K. S., Studies on Polymorphic Modifications of Copper Phthalocyanine. Journal of Solid State Chemistry 2004, 177, (6), 1987-1993. 106. Park, M.; Yoo, H.; Na, S.; Kim, S.; Lee, K.; Friedman, B.; Lim, E.; Iwamoto, M., Phase Transition of Copper(II) Phthalocyanine Thin Films Characterized by a Near-Field Scanning Microwave Microscope. Thin Solid Films 2006, 499, (1-2), 318-321. 107. Usui, H.; Watanabe, M.; Arai, C.; Hibi, K.; Tanaka, K., Vapor Deposition Polymerization of a Polyimide Containing Perylene Units Characterized by Displacement Current Measurement. Japanese Journal of Applied Physics Part I- Regular Papers Brief Communications & Review Papers 2005, 44, 2810-2814. 108. Chen, H. Z.; Ling, M. M.; Mo, X.; Shi, M. M.; Wang, M.; Bao, Z., Air Stable n-Channel Organic Semiconductors for Thin Film Transistors Based on Fluorinated Derivatives of Perylene Diimides. Chemistry of Materials 2007, 19, 816-824. 109. Li, L.; Mizuhata, M.; Kajinami, A.; Deki, S., Different Effects of Alkyl Sulfate and Alkylbenzene Sulfonate Surfactants on the Synthesis and Properties of CuPc/TiO2 Composite Films by the Liquid-Phase Deposition (LPD) Method. Synthetic Metals 2004, 146, 17-27. 152 110. Farag, A. A. M., Optical Absorption Studies of Copper Phthalocyanine Thin Films. Optical & Laser Technology 2007, 39, (4), 728-732. 111. Babel, A.; Wind, J. D.; Jenekhe, S. A., Ambipolar Charge Transport in Air-Stable Polymer Blend Thin-Film Transistors. Adv Funct Mater 2004, 14, (9), 891-898. 112. Leznoff, C. C.; Lever, A. B. P., Phthalocyanine: Properties and Applications. VCH Publisher, InC.: New York, 1989. 113. Evangelisti, M.; Bartolome, J.; de Jongh, L. J.; Filoti, G., Magnetic Properties of α-iron (II) Phthalocyanine. Physical Review B 2002, 66, 144410. 114. Lee, Y.; Tsai, W.; Maa, J., Effects of Substrate Temperature on the Film Characteristics and Gas-Sensing Properties of Copper Phthalocyanine Films. Applied Surface Science 2001, 173, 352-361. 115. Senthilarasu, S.; Sathyamoorthy, R.; Kulkarni, S. K., Substrate Temperature Effects on Structural Orientations and Optical Properties of Zinc Phthalocyanine (ZnPc) Thin Films. Materials Science and Engineering:B 2005, 122, (2), 100-105. 116. Alfonso, E. L.; Tsai, F. Y.; Chen, S.-H.; Gram, R. Q.; Harding, D. R., Fabrication of Polyimide Shells by Vapor Deposition for Use as ICF Targets. Fusion Technology 1999, 35, 131-137. 117. Cunningham, A. F.; Hutchinson, R., Handbook of Radical Polymerization. John Wiley & Sons, Inc: Hoboken, NJ, 2002; p 334. 153 118. Chen, Y.; MaCintyre, D.; Thomas, S., Fabrication of T-shaped Gates using UVIII Chemically Amplified DUV Resist and PMMA. Electronic Letters 1999, 35, (4), 338-339. 119. Gross, S.; Camozzo, D.; Di Noto, V.; Armelao, L.; Tondello, E., PMMA: A key macromolecular component for dielectric low-kappa hybrid inorganic-organic polymer films. European Polymer Journal 2007, 43, (3), 673-696. 120. Tsai, F.-J.; Torkelson, J. M., Microporous Poly(methyl methacrylate) Membranes: Effect of a Low-Viscosity Solvent on the Formation Mechanism. Macromolecules 1990, 23, 4983-4989. 121. Tsai, F.-J.; Torkelson, J. M., Roles of Phase Separation Mechanism and Coarsening in the Formation of Poly(methyl methacrylate) Asymmetric Membranes. Macromolecules 1990, 23, 775-784. 122. Nakanishi, H.; Satoh, M.; Norisuye, T.; Tran-Cong-Miyata, Q., Phase separation of interpenetrating polymer networks synthesized by using an autocatalytic reaction. Macromolecules 2006, 39, (26), 9456-9466. 123. Jansen, B. J. P.; Rastogi, S.; Meijer, H. E. H.; Lemstra, P. J., Rubber-modified glassy amorphous polymers prepared via chemically induced phase separation. 1. Morphology development and mechanical properties. Macromolecules 2001, 34, (12), 3998-4006. 124. Lau, K. K. S.; Gleason, K. K., Initiated Chemical Vapor Deposition (iCVD) of Poly(alkyl acrylates):An Experimental Study. Macromolecules 2006, 39, 3688-3694. 154 125. Jordan, T. E., Vapor Pressure of Organic Compounds. Interscience Publishers, Inc.,: New York, 1954. 126. Indritz, D.; Stone, J.; Williams, F., Vapor Pressure of di-tert-Butyl Peroxide. Journal of Chemical & Engineering Data 1978, 23, (1), 6-7. 127. Wunderlich, W., In Polymer Handbook, Brandrup, J. I., E. H.; Grulke, E., A., Ed. John Wiley & Sons: New York, 1999; p V87. 128. Odian, G., Principles of Polymerization. Fourth ed.; John Wiley & Sons: Hoboken, 2004. 129. Perona, M. J.; Golden, D. M., Very Low-pressure Pyrolysis. VIII. The Decomposition of Di-t-amyl Peroxide. International Journal of Chemical Kinetics 1973, 5, (1), 55-65. 130. Batt, L.; Benson, S. W., Pyrolysis of Di-tertiary Butyl Peroxide: Temperature Gradients and Chain Contribution to the Rate. Journal of Chemical Physics 1962, 36, (4), 895-901. 131. Hiatt, R.; Benson, S. W., Rate Constants for Radical Recombination. IV. Activation Energy for Ethyl Radical Recombination. Journal of the American Chemical Society 1972, 94, (20), 6886-6888. 132. Lee, S. H.; Yoo, P. J.; Kwon, S. J.; Lee, H. H., Solvent-driven dewetting and rim instability. Journal of Chemical Physics 2004, 121, (9), 4346-4351. 133. Reiter, G., Unstable Thin Polymer-Films - Rupture and Dewetting Processes. Langmuir 1993, 9, (5), 1344-1351. 134. Vrij, A.; Overbeek, J. T. G., Rupture of Thin Liquid Films Due to Spontaneous Fluctuations in Thickness. Journal of the American Chemical Society 1968, 90, (12), 3074-3078. 155 135. Brochard-Wyart, F.; Daillant, J., Drying of Solids Wetted by Thin Liquid Films. Canadian Journal of Physics 1990, 60, 1084-1088. 136. Reiter, G., Dewetting of Thin Polymer Films. Physical Review Letter 1992, 68, (1), 75-78. 137. Thiele, U.; Mertig, M.; Pompe, W., Dewetting of an Evaporating Thin Liquid Film: Heterogeneous Nucleation and Surface Instability. Physical Review Letter 1998, 80, (3), 2869-2872. 138. Xu, L.; Shi, T.; An, L., The Dewetting Dynamics of the Polymer Thin Film by Solvent Annealing. Journal of Chemical Physics 2008, 129, 044904-044910. 139. Xue, L. J.; Han, Y. C., Autophobic Dewetting of a Poly(methyl methacrylate) Thin Film on a Silicon Wafer Treated in Good Solvent Vapor. Langmuir 2009, in press. 140. Neto, C., A novel approach to the micropatterning of proteins using dewetting of polymer bilayers. Physical Chemistry Chemical Physics 2007, 9, (1), 149-155. 141. Vourdas, N. E.; Vlachopoulou, M. E.; Tserepi, A.; Gogolides, E., Nano-textured polymer surfaces with controlled wetting and optical properties using plasma processing. International Journal of Nanotechnology 2009, 6, (1-2), 196-207. 142. Ulman, A., Self-Assembled Monolayers of Alkyltrichlorosilanes: Building Block For Future Organic Materials. Advanced Materials 1990, 2, (12), 573-582. 156 143. Elson, J. M.; Bennett, J. M., Calculation of the Power Spectral Density from Surface Profile Data. Applied Optics 1995, 34, (1), 201-208. 144. Janssen, D.; De Palma, R.; Verlaak, S.; Heremans, P.; Dehaen, W., Static Solvent Contact Angle Measurements, Surface Free Energy and Wettability Determination of Various Self-Assembled Monolayers on Silicon Dioxide. Thin Solid Films 2006, 515, (4), 1433-1438. 145. Xing, R. B.; Luo, C. X.; Wang, Z.; Han, Y. C., Dewetting of Polymethyl Methacrylate on the Patterned Elastomer Substrate by Solvent Vapor Treatment. Polymer 2007, 48, 3574-3583. 146. Henn, G.; Bucknall, D. G.; Stamm, M.; Vanhoorne, P.; Jerome, R., Chain End Effects and Dewetting in Thin Polymer Films. Macromolecules 1996, 29, (12), 4305-4313. 147. Reiter, G., Dewetting as a Probe of Polymer Mobility in Thin-Films. Macromolecules 1994, 27, (11), 3046-3052. 148. Reiter, G.; Khanna, R.; Sharma, A., Self-destruction and dewetting of thin polymer films: the role of interfacial tensions. Journal of Physics-Condensed Matter 2003, 15, (1), S331-S336. 149. de Gennes, P. G., Wetting: Statics and Dynamics. Reviews of Modern Physics 1985, 57, (3), 827-863. 150. Morariu, M. D.; Schaffer, E.; Steiner, U., Molecular Forces Caused by the Confinement of Thermal Noise Physical Review Letters 2004, 92, (5), 156102. 157 151. Sharma, A., Relationship of Thin Film Stability and Morphology to Macroscopic Parameters of Wetting in the Apolar and Polar Systems. Langmuir 1993, 9, 861-869. 152. Sharma, A.; Verma, R., Pattern formation and dewetting in thin films of liquids showing complete macroscale wetting: From "Pancakes" to "Swiss cheese". Langmuir 2004, 20, (23), 10337-10345. 153. Williams, M. B.; Davis, S. H., Nonlinear Theory of Film Rupture. Journal of Colloid and Interface Science 1982, 90, (1), 220-228. 154. Mukherjee, R.; Bandyopadhyay, D.; Sharma, A., Control of Morphology in Patterned Directed Dewetting of Thin Polymer Films. Soft Matter 2008, 4, 2086-2097. 155. Seemann, R.; Herminghaus, S.; Jacobs, K., Dewetting Patterns and Molecular Forces: A reconciliation. Physical Review Letters 2001, 86, (24), 5534-5537. 156. Sharma, A.; Mittal, J., Instability of thin liquid films by density variations: A new mechanism that mimics spinodal dewetting. Physical Review Letters 2002, 89, (18), 186101. 157. Reiter, G.; Hamieh, M.; Damman, P.; Sclavons, S.; Gabriele, S.; Vilmin, T.; Raphael, E., Residual Stresses in Thin Polymer Films Cause Rupture and Dominate Early Stages of Dewetting. Nature Materials 2005, 4, 754-758. 158. Sharma, A., Equilibrium and dynamics of evaporating or condensing thin fluid domains: Thin film stability and heterogeneous nucleation. Langmuir 1998, 14, (17), 4915-4928. 158 159. Lide, D. R., CRC Handbook of Chemistry and Physics. 89 Editon ed.; CRC Press/Taylor and Francis: Boca Raton, FL. 160. Ferre-Borrull, J.; Duparre, A.; Quesnel, E., Procedure to Characterize Microroughness of Optical Thin Films: Application to Ion-beam-sputtered Vacuum-ultraviolet Coatings. Applied Optics 2001, 40, (13), 2190-2199. 161. Perry, R. H.; Green, D. W.; Maloney, J. O., Perry's Chemical Engineers' Handbook. 6th ed.; McGraw-Hill: New York, 1984. 162. Liu, Y.; Rafailovich, M. H.; Sokolov, J.; Schwarz, S. A.; Zhong, X.; Eisenberg, A.; Kramer, E. J.; Sauer, B. B.; Satija, S., Wetting Behavior of Homopolymer Films on Chemically Similar Block Copolymer Surface. Physical Review Letters 1994, 73, (3), 440-443. 163. Reiter, G.; Khanna, R., Kinetics of Autophobic Dewetting of Polymer Films. Langmuir 2000, 16, (15), 6351-6357. 164. Wirges, H. P.; Warnecke, H. J.; Friedrich, A., Determination of density, viscosity, and surface tension for the system isobutene-sulfuric acid-tert-butyl alcohol-water. Journal of Chemical Engineering Data 1977, 22, (2), 165-198. 165. Heimenz, P. C.; Rajagopalan, R., Principles of Colloid and Surface Chemistry. Marcel Dekker: New York, 1997. 166. Spencer, N. D.; Moore, J. H., Encyclopedia of Chemical Physics and Physical Chemistry. Taylor & Francis: 2001; Vol. 3. 167. Kwok, A. Y.; Neo, S. A.; Qiao, G. G.; Solomon, D. H., Polymerization-induced Phase Separations in Branched poly(methyl 159 methacrylate) Synthesis. Journal of Applied Polymer Science 2005, 98, (3), 1462-1468.