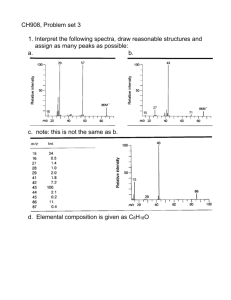
CHAPTER 1 1.1 Introduction
advertisement

1 CHAPTER 1 1.1 Introduction The most efficient organic reactions tolerate unprotected functional groups, generate products in high enantiomeric excess and dramatically increase the molecular complexity in a single step. The ideal situation arises when one can perform multiple bond formation/fragmentation reactions sequentially in one pot. These may be carbon-carbon, carbon-heteroatom or heteroatom-heteroatom bond manipulations. The difficulties involved in designing these ideal reactions are considerable, with high selectivity and high yield of each reaction being imperative. The efforts of good design are, however, well rewarded by a successful reaction sequence due to the limited isolation and purification steps that would otherwise be necessary. As the number of overall reactions within a sequential reaction process increases, its complexity rises almost exponentially and one finds that only a select group of reagents is able to face up to the considerable task. One such reagent capable of sequential processes is samarium diiodide (SmI2). 1 The reagent was first discovered in the late 1970s and has dramatically changed the field of organic synthesis. A large part of samarium diiodide’s potency stems from the fact that it is capable of promoting both radical and anionic chemistry. Examples of these processes include radical cyclisations, ketyl-olefin coupling reactions, pinacol coupling reactions, Barbier-type reactions, aldol-type reactions, conjugate additions, and nucleophilic acyl substitutions. 2 The versatility of SmI2 can be further enhanced by the addition of catalysts,1a by solvent effects 2 or through other variations of other reaction conditions. 3 The ability to fine tune the activity of the SmI2 allows it to be used under both sensitive and stringent conditions. Depending on the type and rate of addition and any additives employed, samarium diiodide can promote both one and two electron processes or a combination of the two. These may be sequential radical processes, first radical and then anionic processes, tandem anionic processes or carbanionic reactions followed by radical reactions. 1.2 Individual Reactions Promoted by Samarium(II) Iodide 1.2.1 Single-Electron Processes There are a number of different functional groups that SmI2 can reduce to generate radicals. Alkyl, aryl and alkenyl radicals can be produced from halide substrates while ketyl radical anions are produced from aldehydes and ketones. The rate and selectivity of the different reductions depend on the type of halide used and are often substrate dependent as well. 1.2.1.1 Alkyl, Aryl and Alkenyl Radical Reactions The initiation of the radical reactions usually takes place by the reduction of organic halides. The intrinsic limiting factor in this process is that SmI2 can further reduce the radical formed to the corresponding carbanion (Scheme 1). Thus, any desired radical 3 reaction (krad) must take place significantly faster than the reduction to the anion (kred). This reactant is often employed instead of tin hydride and silicon hydride reagents. A distinct advantage of this approach is the fact that an organosamarium species may be formed if necessary for a sequential process. For a range of alkyl halides, reactivity of the substrate decreases in the order: I>Br>Cl.1a SmI2 R-X SmI2 kredn R-SmI2 E+ R-E 3 4 R 2 1 'R SmI2 krad 5 (reaction) 'R-SmI2 E+ 'R-E 6 7 Scheme 1 Radical processes promoted by SmI2 will only be synthetically useful if the radical has a sufficient lifetime to undergo the desired radical process. Although many intramolecular processes exist that meet this requirement, there is only a single class of intermolecular process for which this rings true, namely the SmI2-promoted radical chain coupling of polyhaloalkanes to alkenes and alkynes (Scheme 2). 4 F F SmI2 + Cl(CF2)2I 8 Cl THF, 80% 9 10 Scheme 2 I F F 4 The most common form of radical process is radical cyclisation. Of the more useful types of reactions are the cyclisations of 6-halohex-1-ynes (Scheme 3). 5 The reduction of the halo atom produces a non stabilised alkyl radical intermediate, which then cyclises onto the alkynyl moiety to give the cyclopentylidene radical. This then abstracts an hydrogen atom from the solvent or another suitable donor. It should be understood that radicals such as aryl or alkenyl radicals abstract donor atoms from their surrounds faster than SmI2 can reduce them. 6 This is an important consideration to take into account when attempting a sequential process. R R X 3 SmI2 R R SH +S THF, DMPU 12 11 13 14 15 Scheme 3 Substrates containing an oxygen atom within the chain are generally reduced more rapidly than their all-carbon analogues. This phenomenon has been exploited in the synthesis of lactones (Scheme 4). 7 n-Pr Br BnO n-Pr 1. 2.5 SmI2 O THF, HMPA, t-BuOH 2. H2CrO4 16 O O 17 Scheme 4 5 Another useful type of alkyl radical can be derived from N-[(N’,N’-dialkylamino)alkenyl]benzoytriazoles (Scheme 5). Mixtures of diastereomers are usually obtained in these reactions, with the cis isomers generally predominating. 8 O O SmI2 N EtO2C N CO2Et THF N N 18 19 N Scheme 5 The formation of alkyl radicals is not limited to halogens, sulfones and related functional groups, but can be formed by reductive cleavage of cyclopropyl ketones via a rearrangement process (Scheme 6). The resultant radical is usually trapped intramolecularly producing bicyclic systems. 9 These cleavage reactions will be dealt with in more detail in a later section (see Chapter 1.5.2.2). O O SmI2 TMS THF, HMPA 20 TMS 21 Scheme 6 Although alkenes and alkynes are the most commonly used radical acceptors, some workers have made use of hydrazones. Despite both SmI2 and tin hydride having been 6 utilised for the cyclisations of halohydrazones to cyclopentylhydrazines, 10 SmI2 is the preferred reagent giving better diastereoselectivies at lower temperatures (Scheme 7). X NNPh2 H NNPh2 4.5 SmI2 H NNPh2 + THF, HMPA 23 22a: X = Br 22b: X = I 7:1 ds 24 11: 1 ds Scheme 7 The observed diastereoselectivity can be rationalised by viewing the two possible transition state chair conformations. The structure leading to a trans product suffers from an unfavourable 1,3-diaxial interaction between the substituent at the radical centre and one of the hydrogen atoms on the ring, while this high energy species is absent in the chair conformation of the cis product’s transition structure (Scheme 8). H N NPh2 H R 25 Disfavoured H N NPh2 R H 26 Favoured Scheme 8 Benzofurans, naphthofurans and indoles have been synthesised using aryl radical methodology.2d,6 The reactions proceed smoothly in the presence of tetramethylguanidine (TMG), which seems to be a superior additive compared to HMPA or DMPU for these types of reactions (Scheme 9). 7 Br SmI2 N N THF, HMPA 28 27 Scheme 9 1.2.1.2 Ketyl Radical Anion Reactions The ketyl radical-olefin coupling reaction is to date the most studied of all the different processes that samarium(II) iodide promotes. The coupling serves as an highly useful procedure to couple aldehydes and ketones to alkenes and alkynes. The intermolecular version, although popular, has been used less frequently than its intramolecular counterpart. This is due mainly to the fact that the reaction is restricted to alkenes or alkynes that are good electrophiles. The products of these reactions are their corresponding lactones (Scheme 10). 11 CHO + 29 CO2Et 2 SmI2 THF, t-BuOH 30 O O 31 Scheme 10 In addition to conjugated esters and nitriles, other conjugated alkenes such as styrene, alkenylsilanes, vinyl acetates and allylic acetates serve as efficient ketyl radical acceptors in SmI2-promoted intermolecular coupling reactions. 12 The reactions with unactivated 8 alkenes are, however, not as selective as the activated ones and often lead to mixtures of isomers (Scheme 11). O + Ph OH 2 SmI2 OTMS 32 33 THF, HMPA t-BuOH Ph O 34 Scheme 11 Alkynes also serve as suitable acceptors for ketyl radicals in intermolecular coupling reactions (Scheme 12). 13 The carbon-carbon bond formation always takes place at the terminus of external alkynes and at the most electropositive carbon atom of internal alkynes. An activating group is preferential in obtaining suitable yields. O + Ph 35 OH 2 SmI2 Ph TMS 36 THF, HMPA t-BuOH Ph TMS 37 Ph Scheme 12 More commonly employed are the intramolecular ketyl olefin coupling reactions. These are quite efficient and a wider range of acceptors is tolerated. This methodolgy has been used to synthesise a number of ring structures varying in size from strained three membered rings to larger eight membered ring systems. Chelation can be used to control the relative stereochemistry of the hydroxyl and carboxylate stereocentres formed during the cyclisation.6b Chelation of the two carbonyl groups, by the Sm(III) Lewis acid generated, provides a template for stereochemical 9 control, providing a facial bias in the attack of the ketyl radical onto the unsaturated ester (Scheme 13). O O HO OEt EtO2C CO2Et 2 SmI2 THF, t-BuOH CO2Et 39 38 Scheme 13 The proximity of hydroxyl groups within the substrate has a significant influence on the stereoselectivity of the reaction. 14 Even in the absence of additives such as HMPA, these reactions proceed smoothly at low temperature with good yields and high diasteroselectivity (Scheme 14). The addition of HMPA actually decreases the stereoselectivity, thus demonstrating that the chelation of the Sm(III) species by the ketone and hydroxyl groups plays a significant role. OH OH SmI2 THF, MeOH O CO2Me OH CO2Me 41 40 Scheme 14 Although the carbonyl olefin coupling to form carbocycles is the most widely used protocol, activated alkenes have been employed in the preparation of nitrogen 10 heterocycles (Scheme 15). 15 The yields of these reactions are, however, low and a mixture of isomers is isolated. O N MeO OHC O OPh OTBS 2 SmI2 N THF, HMPA MeO t-BuOH HO OPh TBS 43 42 Scheme 15 The radical addition-elimination reaction of ketones with allyl sulfides has been investigated in the synthesis of (-)-grayanotoxin III.16 The diasteroselectivities are quite high although the observed olefin geometries are not stereospecific, when applicable. The reaction can also be carried out with allyl sulfones (Scheme 16). BnO Y 3 SmI2 CHO THF, HMPA 44a: Y = SPh 44b: Y = SO2Ph OH BnO 45 Scheme 16 The coupling reactions are not limited to alkenes and similar results have been achieved using activated alkynes. Carbocycles, nitrogen heterocycles and oxygen heterocycles have been prepared in this way (Scheme 17). 17 11 TMS O N t-Boc 2 SmI2 TMS HO THF, HMPA t-BuOH N t-Boc 47 46 Scheme 17 The ketyl radical is distinguished from its alkyl radical counterpart by the fact that it can undergo cyclisation onto unactivated alkenes, alkynes and other functional groups. The 5exo-trig cyclisation represents one of the simplest of these transformations. The diastereoselectivity generally varies with the type of substituent next to the ketone (Scheme 18). 18 O 2.2 SmI2 HO R THF, HMPA t-BuOH R 48a: R=Me 48b: R=Ph HO R + 49 150:1 1:150 50 Scheme 18 The outcome of the reaction can be rationalised on the basis of a chair-like transition structure (Scheme 19). The favoured state 51 predominates due to electronic effects as long as the substituent R is small. As the steric bulk of R increases the disfavoured state 52 becomes favourable due to axial-axial interactions between R and the methylene group in 51. 12 OSmI2-Ln OSmI2-Ln R R H Disfavoured Favoured 51 52 Scheme 19 The 5-exo-trig bicyclisation reactions promoted by SmI2 generally occur with high diastereoselectivities. Both linearly fused bicyclics (Scheme 20)13 and bridged systems (Scheme 21) have been synthesised using this methodology. 19 O Et 2.2 SmI2 HO THF, HMPA t-BuOH 53 54 Scheme 20 O 2.2 SmI2 OH H THF, HMPA t-BuOH Me 56 55 Scheme 21 Similarly, alkynes have also been chosen to be radical acceptors (Scheme 22).17 Yields are modest for unactivated alkynes but can be improved by activating them with substituents such as trialkylsilyl groups. Although ketone ketyl precursors seem to 13 provide higher yields than their aldehyde counterparts, an aldehyde/alkyne coupling has been used in the synthesis of isocarbacyclin. 20 HO CHO 2.2 SmI2 C5H11 TBDMSO THF, HMPA t-BuOH TBDMSO C5H11 OH OH 58 57 Scheme 22 The coupling of allenes is also possible, although only a limited number of reactions has been studied (Scheme 23). 21 O O 2 SmI2 MeO O MeO + CHO THF, t-BuOH OH 60 59 MeO OH 2:1 61 Scheme 23 Fewer studies of 6-exo cyclisation have been carried out as they suffer from lower yields and selectivities (Scheme 24).18 The scope of these reactions is similar to those discussed in the 5-exo-trig cyclisation case. They include cyclisation of the ketyl radical onto alkenes, alkynes and allenes to form both cyclic and bicyclic structures. 14 O 2.2 SmI2 HO THF, HMPA t-BuOH 62 63 17:1 ds Scheme 24 Pinacol coupling reactions of ketyl radicals have been well studied. The reactions of aromatic compounds are highly substrate dependant as well as sensitive to the reaction conditions and any additives employed. 22 The treatment of benzaldehyde with SmI2, for example, leads to an almost quantitative yield of the corresponding pinacol product. Upon repeating the reaction in the presence of HMPA, however, the aryl coupled product is obtained in good yield with hydrobenzoin being the minor isomer (Scheme 25). This observation is consistent with the notion that successful pinacol coupling reactions require simultaneous complexation of both carbonyl moieties to one Sm ion. CHO SmI2 OH + CHO HO OH 66 65 64 THF 0% 100% THF/HMPA 60% 10% Scheme 25 The coupling of non-aromatic substrates is much simpler. In the absence of a proton source such as methanol, aldehydes and ketones are easily coupled in the presence of SmI2 to their corresponding pinacol products (Scheme 26). 23 If a proton source is indeed 15 present, the carbonyl group undergoes simple reduction to give the respective alcohols. n-C6H13C(O)CH3 OH SmI2 THF 67 n-C 6H13 n-C 6H13 OH 68 Scheme 26 In the same family of reactions is the coupling of aldimines using SmI2 (Scheme 27). 24 Both alkyl and aryl aldimines can be coupled. SmI2 PhCH NPh THF 69 NHPh Ph Ph NHPh 70 Scheme 27 Cyclisation reactions of 1,5- and 1,6-dialdehydes or diketones using the pinacol methodology proceed smoothly with considerable stereochemical control: the cis-diols are almost exclusively obtained (Scheme 28). 25 Heteroatom functionality α to the carbonyl group is tolerated showing that pinacolisation is much faster than reductive cleavage. Furthermore, polar substituents α to the carbonyls end up anti to the diol stereocentres in the final product. OBn TBDMSO CHO O OBn SmI2 OH THF OH TBDMSO 71 72 Scheme 28 16 Although ketyl radical/nitrile coupling is possible, the coupling process is slow and the yield is low (Scheme 29). This is indicative of the reluctance of the nitrile moiety to undergo radical addition.6b,26 O O OH CO2Et SmI2 OEt THF, HMPA NC 73 O 74 Scheme 29 A somewhat better radical acceptor is the hydrazone moiety. Cyclisation of carbonylhydrazones provides an excellent route to 2-aminocyclopentanols (Scheme 30).10 N NPh2 H O n NHNPh2 SmI2 R R THF, HMPA n 75 R = H,Me n = 1,2 OH 76 Scheme 30 1.2.2 Two-Electron Processes The ability of samarium diiodide to promote both one- and two-electron processes sets it apart from most other reductive coupling reagents presently available. The two-electron process complements the more common organolithium, organomagnesium and organozinc chemistry. 17 1.2.2.1 Barbier- and Grignard-Type Reactions The samarium Grignard reaction is limited in scope to primary and secondary halides and even then the reaction does not match up to its organolithium and organomagnesium counterparts. The samarium Barbier reaction is just the opposite, succeeding where the magnesiumand lithium-promoted reactions fail. Samarium diiodide promoted reactions also have a broader scope and tolerate a wider range of functionalities. Although the intermolecular Barbier reaction is limited in use, the intramolecular form is widely used and has found an important niche in organic synthesis. Allylic, propargylic and benzylic halides are highly reactive in samarium Barbier reactions, with both aldehydes and ketones serving as partners for the reactions (Scheme 31).26 HO CHO I + 2 SmI2 THF 77 78 79 Scheme 31 The active functionality can be extended to sulfones. These reactions require HMPA as an additive to ensure a successful reaction (Scheme 32). 27 18 SO2Ph + n-C 6H13CHO OH 4 SmI2 n-C 6H13 THF, HMPA 81 80 82 Scheme 32 Selective transformations of polyfunctional substrates can be accomplished by taking advantage of the difference in reduction potentials between alkyl iodides, bromides and chlorides (Scheme 33).1a 2 SmI2 I(CH2)6Cl + n-C 6H13C(O)CH3 83 THF HO n-C 6H13 84 (CH2)6Cl 85 Scheme 33 The most promising of the samarium Barbier reactions are the intramolecular processes that form five or six membered rings upon cyclisation. These reactions allow easy access to cyclopentanol 28 and cyclohexanol 29 derivatives (Scheme 34). O Br 2 SmI2 OH THF 86 87 Scheme 34 The same protocol has been applied to diverse systems and in virtually every case has been superior to conventional methods. Suginome and Yamada successfully employed this technique in the synthesis of (±)-exaltone and (±)-muscone (Scheme 35). 30 19 O HO 2 SmI2 I H THF, HMPA 89 88 Scheme 35 Another use of the reaction is that of making bridged bicyclic alcohols (Scheme 36). 31 The methodology has been applied to make highly strained and complex molecules that are otherwise accessible only with some difficulty. O OH 2 SmI2 THF, cat. Fe(III) I 90 91 Scheme 36 1.2.2.2 Aldol- and Reformatsky-Type Reactions The Reformatsky reaction promoted by samarium(II) iodide has been carried out between α-halo esters and ketone electrophiles (Scheme 37).1a, 32 O CO2Et 2 SmI2 HO CO2Et + Br 92 THF, HMPA 93 94 Scheme 37 20 Similarly aldol-type reactions between α-halo ketones and suitable aldehydes have proven successful (Scheme 38). 33 O Br + O2N CHO THF, HMPA 95 O 2 SmI2 96 Ph NO2 97 Scheme 38 Condensations between α-keto carboxylates and α-halo ketones have also been attempted with good yield. 34 The intramolecular reaction, as before, finds more use in organic synthesis. Reactions of SmI2 with β-bromoacetoxy carbonyl substrates are proposed to generate a Sm(III) ester enolate with cyclisation taking place through a rigid cyclic transition structure enforced by chelation (Scheme 39). 35 O Br H O O SmI2 R1 R3 R2 H 98 R3 R2 H R1 R2 R1 OH O O 99 Scheme 39 O Sm(III) R3 O 100 O 21 1.2.2.3 Nucleophilic Acyl Substitution Reactions Although unreactive in intermolecular reactions, esters and amides undergo highly selective nucleophilic acyl substitution reactions when appropriately substituted carboxylic acid derivatives are treated with SmI2 (Scheme 40). 36 The method is amenable to the synthesis of four-, five- and six-membered rings with primary alkyl, secondary alkyl and allylic halides participating in the reaction. O O I 2 SmI2 Y Ph Ph THF 101a: Y = OEt 102 101b: = S(i-Pr) 101c: = N(OMe)Me 71% 68% 81% Scheme 40 1.3 Sequential Reactions Promoted by Samarium(II) Iodide The ability of SmI2 to promote sequential processes has been a major factor contributing to its popularity as a reducing agent in organic synthesis. Another extremely useful property of the reagent is that the individual reactions within a reaction sequence may be either one- or two-electron processes. As the sequential processes are just a combination of the above mentioned individual chemistries, only a limited number of examples shall be covered for each set. 22 1.3.1 Sequential Radical Processes The success of tin hydrides and silicon hydrides in promoting sequential radical processes has overshadowed SmI2 as a potent reagent for the above and thus very few examples of SmI2-mediated sequential radical processes have been reported. One such example has been employed in the key step in the synthesis of (±)-coriolin (Scheme 41). 37 CHO O 1. 1.3 SmI2, HMPA O 2. p-TSA, acetone HO H H HO H H O H 103 91% O + + H 104 105 OH O O 106 Scheme 41 1.3.2 Radical/Anionic Sequences These are the most widely studied and most commonly used sequential processes. Generally, a radical intermediate is formed after SmI2 has reduced a suitable precursor. The radical intermediate then undergoes an addition onto a radical acceptor forming a second radical intermediate. This is then further reduced by a second equivalent of SmI2 to form the organosamarium that can be trapped by a number of electrophiles (Scheme 42). 38 Only primary or secondary radicals can be reduced to the organosamarium with tertiary radicals abstracting an hydrogen atom or undergoing disproportionation.38 23 I 1. 2 SmI2 O O 2. 3-pentanone OH 108 107 Scheme 42 1.3.3 Anionic/Radical Sequences The anionic/radical process is characterised by the addition of four or more equivalents of SmI2. The first two equivalents are responsible for producing the organosamarium that normally undergoes a nucleophilic acyl substitution reaction. The resulting ketone is then reduced by one equivalent of the remaining SmI2 and further undergoes a radical cyclisation process. Subsequent reduction of the radical and protonation yields the desired multistep product (Scheme 43).36,39 The sequence can be repeated by quenching with a suitable electrophile instead of a proton source. O I OEt 4 SmI2 O HO THF, HMPA 109 110 111 Scheme 43 1.3.4 Sequential Anionic Processes The most straightforward reaction of this type is one in which two intramolecular Barbier type reactions occur in one pot. Although Molander has classed this particular reaction as 24 sequential, it is clear that two separate reactions occur at two different centers. This strategy has been used in the synthesis of polyquinenes (Scheme 44). 40 Br H 4 SmI2 O O THF, HMPA OH HO H Br 113 112 Scheme 44 1.4 HMPA - Effects and Mechanistics The fact that Sm(II) is multivalent suggests that the redox potential of a divalent Sm species will vary depending on the number and type of ligands coordinated to it. Although HMPA 41 is the most widely used co-solvent in SmI2 mediated reactions, other co-solvents containing basic oxygen including 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)pyrimidinone (DMPU) 42 and 1,1,3,3-tetramethylurea (TMU) 43 have been used with success. 1.4.1 The Effect of HMPA on the Reduction Potential of SmI2 Although there were numerous synthetic examples of the ability of HMPA to enhance the yield and selectivity of a SmI2 mediated reaction,1 it was not until the work of Flowers that this effect was quantitatively studied. 44 25 The change in redox potential of a reducing agent may significantly alter the mechanistic pathway that a reaction follows, possibly with deleterious effects. The determination of the redox potentials of a series of Sm(II) species is thus of utmost importance when designing a reaction sequence. Species that do not succumb to SmI2 in THF may be readily reduced within a SmI2-HMPA system. One way in which to determine the energetics of an one-electron reducing system is to make use of electrochemistry. Flowers studied the effect of HMPA by adding aliquots of HMPA to a SmI2-in-THF system and subsequently recording a linear sweep voltammogram for each co-solvent addition (Table 1). Only a slight change in the reduction potential is observed after the addition of up to two equivalents of HMPA. Entry Equivalent of Oxidation ΔE, V (kcal) HMPA vs. SmI2a Potential Vb 1 0 -1.33 0 2 1 -1.43 0.10 (2.3) 3 2 -1.46 0.13 (3.0) 4 3 -1.95 0.62 (14.0) 5 4 -2.05 0.72 (16.6) 6 5 -2.05 0.72 (16.6) 7 6 -2.05 0.72 (16.6) a) conc. of SmI2 = 0.5 mM. B) vs. Ag/AgNO3 reference electrode in THF. Table 1 26 However, on addition of the third equivalent, one sees a more drastic increase in the reducing power of the SmI2-HMPA species from –1.46 V to –1.95 V. On addition of the fourth portion, one again sees a significant (although less so) increase but this plateaus on the addition of five or more equivalents. Hou and co-workers 45 have successfully isolated and obtained crystallographic data of a SmI2(HMPA)4 complex which they believed to be the reactive species in the SmI2HMPA mediated reaction. This is consistent with the above electrochemical investigation. Another validation of these results comes from the work of Curran. 46 He investigated the rate constants of the reactions of primary alkyl radicals promoted by SmI2 with varying HMPA concentrations, and observed that the reaction time of 6-iodo1-hexene was too slow for their rate experiments in the absence of HMPA. Upon addition of 2 equivalents of HMPA the rate became measurable and reached a maximum on addition of the fifth equivalent. 1.4.2 A Structural Basis for the HMPA Effects HMPA is not only responsible for increasing the reduction potential of SmI2 but also influences the regio- and stereo-selectivity of SmI2-promoted reactions. 47 Despite the extensive use of the SmI2-HMPA complex in organic synthesis, the exact role played by HMPA in these reactions was not well understood prior to the isolation and structure determination of the SmI2-HMPA complex by Hou and co-workers. 48 They were posed with the following questions: a) Why does HMPA increase SmI2’s reducing ability? b) Why is the regio- and stereochemistry of the reaction altered upon addition of HMPA? c) 27 How much HMPA is necessary for optimum results? d) Why does the selectivity become poorer when the HMPA exceeds a certain amount? To get a better understanding of the concepts involved in these effects it was necessary to isolate and structurally characterise various SmI2-HMPA complexes. [SmI2-(HMPA)4] The single crystal was obtained by adding 4.5 equivalents of HMPA to a solution of SmI2 in THF, and subsequently crystallising it from toluene (HMPA binds to the metal through the oxygen atom on the phosporus). The structure reflected a slightly distorted octahedral complex (Figure 1), the most notable feature of which was the short Sm-O(HMPA) bonds (av. 2.500 Å) whereas other oxygen-donor ligands have longer SmO bond distances (av. 2.600 Å). 49 Due to the strong co-ordination of the four HMPA ligands the Sm-I bonds were also much longer (av. 3.390 Å) than those found in other SmI2 complexes such as cis-[SmI2(dme)(thf)3] (av. 3.246 Å). This was indicative of weaker Sm-I bonds. The four HMPA ligands as well as the iodides were bound in the innersphere. SmI2 + HMPA 1 : 4.5 THF I HMPA HMPA Sm HMPA HMPA I 114 Figure 1 SmI2-(HMPA)6] The complex was isolated after the addition of 10 equivalents of HMPA to a solution of SmI2 in THF. The excess was added in order to establish the maximum 28 number of ligands that would co-ordinate to the Sm(II) ion. The complex was also octahedral in shape but featured the iodide ions in the outer sphere position (Figure 2). Due to the six bulky HMPA ligands, the Sm-O bonds in this complex were slightly longer than before (av. 2.531 Å) but still considerably shorter than those between Sm(II) and other oxygen-donor ligands. 2+ SmI2 + HMPA 1 : HMPA THF HMPA 10 HMPA HMPA Sm HMPA HMPA - 2I 115 Figure 2 Based on the information presented by the crystal structures, it becomes clear that complexation of HMPA to the Sm(II) ion greatly changes both the electronic and steric environments around the metal. The long Sm-I bond distance suggests that the bond is relatively weak and is easily cleaved by another co-ordinative ligand. The Sm(II) metal centre will accept as many as six strongly electron-donating ligands when a sufficient amount of HMPA is added. This would explain the enhancement of the reduction potential of SmI2 upon addition of HMPA. When four equivalents of HMPA are added a square planar co-ordination geometry of the ligands around the metal ion is adopted, which has been used to explain the selectivity observed in many SmI2-mediated reactions. It is proposed that a substrate will approach the metal centre from either above or below the square plane formed by the HMPA 29 ligands. The steric repulsion between the substrate and the bulky ligands will naturally change the regio- and stereoselectivity of the reactions as compared to those carried out in the absence of HMPA.47 When six or more equivalents of the co-ordinating ligand are used, the Sm-I bonds are cleaved and the metal centre is surrounded by six bulky HMPA ligands. In this scenario the approach of substrates is blocked and the electron-transfer reaction takes place in the outer sphere. The study shows that if one is interested only in increasing the reducing power of SmI2, the use of six or more equivalents of HMPA is appropriate. If, however, one would like to control the selectivity of a reaction, only a limited amount of the ligand should be added. Although most information concerning the structure of both the Sm(II) and Sm(III) oxidation states has been provided through crystallographic studies, little work has been done on the structure of complexes of Sm and HMPA in solution. Flowers and coworkers have studied the structure and energetics of the samarium diiodide-HMPA complex in THF using UV-vis spectroscopy, isothermal titration calorimetry and vapor pressure osmometry. 50 . They found that the aggregation number for SmI2 in THF was 0.98 ± 0.09 over the entire concentration range studied, indicating that SmI2 is monomeric as well as the fact that the addition of HMPA cosolvent to a solution of SmI2, displaces the THF bound to the metal. If four equivalents of HMPA are added the iodide ions remain 30 innersphere while on the addition of six or more equivalents of HMPA forces the iodide ions to an outersphere position. Crystal structures of Sm(III) complexes indicate an octahedral environment about the metal centre. 1.5 Carbon-Carbon Bond Fragmentation Reactions 1.5.1 C-C fragmentation reactions in ‘no-strain’ systems Although various dehalogenation and deoxygenation reactions of α-hydroxy, α-alkoxy, or α-acyloxycarbonyl compounds utilising samarium(II) iodide have been extensively investigated, comparatively little work has been done on C-C bond fragmentation reactions. Magnus and co-workers published one of the first C-C bond fragmentation reactions promoted by SmI2 (Scheme 45). 51 The reagent was resorted to after a reaction with Bu3SnH in toluene (40 h at reflux) yielded a mixture of the desired fragmentation product and the product of simple reduction in modest yield. Upon subjecting the steroid derivative to SmI2 in THF at room temperature, the C-C cleaved product was isolated in 88% yield in only five minutes, and none of the reduced product was detected. The authors suggested that a plausible explanation for the success of the reaction using SmI2 where the tin reagent failed was the ability of the Sm(II) to rapidly reduce a radical to an anion. The radical mechanism can, however, not be discarded. 31 O MeS S O SmI2 H H O H O H H H + THF H Me O H O HO 116 117 118 88% 0% Scheme 45 Honda and co-workers 52 have successfully prepared physiologically active, chiral, natural products using a carbon-carbon bond cleavage reaction as the key step. Starting from a chiral cyclopentane derivative, they were able to regioselectively cleave the α,β-C-C bond with respect to the ester functionality (Scheme 46). Since SmI2 can complex both the halogen atom as well as the carbonyl moiety γ to the Cl, forming a seven-membered ring transition state, a concerted fragmentation mechanism was proposed in converting the carbocycle into the acyclic alkene. The reaction was successful using both the bromo and chloro substrates. OTES OTES 3 SmI2 Cl CO2Me CO2Me THF, HMPA 86 % 119 - OTES OMe Cl O [Sm2+] 121 Transition State Scheme 46 120 32 In the absence of HMPA, none of the fragmented product was isolated: reductive dehalogenation took place in good yield. The reactions were proved to be SmI2 specific with no fragmentation taking place under other standard reducing conditions. These included the treatment of the ester with tri-n-butyltin hydride and azobisisobutyronitrile (AIBN) in refluxing benzene under radical-initiating reaction conditions, and the reduction of the substrate with zinc powder in acetic acid. The authors were cautious in proposing a mechanism for the observed fragmentation. However, they speculated that the results suggest that the reaction would occur via a twoelectron reduction process, involving further reduction of the initially formed radical anion prior to either proton abstraction or a radical-induced fragmentation (see transition state above). Reaction of the ketone analogue of the ester, i.e. the γ-halo ketone, also yielded the bond cleaved product. In contrast, treatment of open-chain γ-halo ketones (halogen = Cl, Br) with SmI2 under the same conditions has been known to yield cyclobutanols, via the ketyl radical. 53 The structural bias or inherent strain in forming a bicyclo[3.2.0]heptane ring system is probably the deciding factor which promotes the fragmentation over cyclisation. Honda has used the SmI2 promoted fragmentation of γ-halo esters for the synthesis of a number of enantiomerically pure natural compounds including alkaloids (Scheme 47), terpenes and antibiotics. 54 33 OTBDMS Cl CO2Me O 122 N H 123 Scheme 47 The fragmentation reaction of an ε-halo-α,β-unsaturated ester, where a similar cleavage reaction involving the fragmentation of the carbon-carbon bond between the γ and δ positions of the carbonyl group, was also investigated (Scheme 48). 55 The substrate underwent bond scission at room temperature upon treatment with SmI2 / HMPA to give a 2:1 ratio of stereoisomers (E:Z), in 91% yield. OTES OTES SmI2 O O THF, HMPA Cl O CO2Et CO2Et 125 124 OH OH Nemorensic acid 126 Scheme 48 Molander has described stereocontrolled cyclisation reactions of a number of substrates mediated by SmI2. 56 Intramolecular cyclisation of allylic halides of varying chain lengths onto β-keto esters successfully provided vinyl-substituted cyclopentanes and cyclohexanes in good yields as a mixture of isomers. However, the attempted cyclisation of ethyl 2-methyl-2-(trans-4-bromo-2-butenyl)-3-oxobutanoate produced ethyl 2-methyl3-oxobutanoate instead of the desired cyclobutane or cyclohexane derivative. The authors 34 suggested that loss of butadiene as required for this transformation is facilitated by the ability of a β-keto ester stabilised anion or radical intermediate to serve as an effective leaving group in the reaction (Scheme 49). This result implied that the rate of fragmentation is substantially faster than that of cyclisation, which would normally first require reduction of the radical intermediate to the corresponding anion. O O Me O OEt Me Br 2 SmI2 O OEt Me Me THF, - 78 °C 128 127 Scheme 49 This theory is supported by the outcome of the reaction shown below (Scheme 50). In this case, fragmentation would result in a less stabilised anion/radical species than that of the previously mentioned reaction; treatment of trans-8-bromo-4-methyl-6-octen-3-one with SmI2 afforded 1-ethyl-6-methyl-3-cyclohexen-1-ol in 91% isolated yield. O Me Me Br 2 SmI2 Et OH Me THF, - 78 °C 129 130 Scheme 50 Samarium(II) iodide has been successfully used to promote reductive decyanation of malonitrile derivatives (Scheme 51). 57 The decyanation was achieved using a range of 35 either monosubstituted (R1 = alkyl, R2 = H) or disubstituted (R1 = alkyl, R2 = alkyl) malonitriles in good yield. It was found that HMPA was essential for the success of the reaction. In the substrates where one of the alkyl groups contained C-C unsaturation, no cyclisation occurred where either a five- or seven membered ring could have formed. The authors expanded on this work and were able to apply this new methodology to decyanate α-alkoxycarbonyl substituted nitrile derivatives. Again, the addition of HMPA was critical for the reaction pathway to proceed. Although Bu3SnH can be used for the decyanation of malonitrile derivatives, the decyanation of the α-alkoxycarbonyl nitrile compounds seemed to be SmI2 specific. R2 CN SmI2, THF R2 CN R1 CN HMPA, 0 °C R1 H 131 132 R2 CO2Et SmI2, THF R2 CO2Et R1 CN HMPA, rt R1 H 134 133 Scheme 51 Although several methods of producing macrocyclic lactams have been forthcoming, the construction of these large ring systems still presents a significant challenge because ring closure is difficult to achieve. 58 With the discovery that alkylazides are reactive towards reduction by SmI2 came an investigation into the ring enlargement of readily available azidocyclododecanones to large-ring lactams (Scheme 52). 59 Although the exact mechanism of the reaction is unknown, the synthetic route is mild yet efficient in 36 producing 16- and 17-membered lactams. Attempts to synthesize larger ring systems by this route failed. O CN N 3 n O O H N CN SmI2, THF NH NH + rt, 2 h, 92% (2:1) n=3 135 136 137 Scheme 52 1.5.2 C-C fragmentation reactions in ‘ring-strained’ systems 1.5.2.1 Cyclobutane containing substrates A more common form of C-C cleavage is that of strained ring systems, including compounds that contain a cyclobutane moiety. The initial fragmentation of the following cyclobutane substrate led to an allylic radical, which upon further reduction and protonation yielded an isomeric mixture of fragmented products (Scheme 53). 60 The exact ratio of the mixture was shown to be dependent on the nature of the reducing agent employed. The reagents used were n-Bu3SnH, (C6H5)3SnH and SmI2, with AIBN used as the initiator for the former two reagents. 37 CH2I 2 SmI2 + 93 % R 99 R = H, Me, CO2R' R' = Me, t-Bu 139 138 R 1 R 140 Scheme 53 With the hydride reagents the exocyclic double bond isomer predominated while with SmI2 the endocyclic double bond product was favoured. It was established that activation of the double bond was not necessary for the success of the reaction as displayed by the two cases where R = H and R = Me. SmI2 was determined to be the reagent of choice in these transformations with the fragmentation yields being in excess of 90 % in all four cases. The reactions were assumed to proceed via a radical pathway forming the allylic radical after fragmentation of the strained cyclobutylcarbinyl system (Scheme 54). This radical was then reduced to the carbanion by a second equivalent of SmI2, and finally protonated. Support of this theory is provided by the fact that quenching with excess MeI gave only the α-methylated product when making use of the ester substrates, which, at the very least, proves that the reaction ends in a carbanion. 38 CH2I SmI2 SmI2 CO2Me CO2Me 141 142 CO2Me 143 MeI Me CO2Me 144 Scheme 54 Fragmentation of appropriate [2+2] photoadduct derivatives led to bicyclo[m.n.0]carbon skeletons that are present in a wide range of natural products. This methodology has been used in the synthesis of Dictamnol, a trinor-guaiane (Scheme 55). 61 The key step involves the initial reduction of a diiodo compound and a subsequent free radical fragmentation. Reduction of one iodo moiety initiates the reaction while the other iodide serves as a leaving group in the last stage of the fragmentation sequence. Treatment of the substrate with SmI2 in THF and DMPU provided the ring expanded 5,7 fused ring diene in good yield. Apart from the generally higher yield offered by SmI2, this reagent is preferable to n-Bu3SnH as the radical initiator because of the convenience, lower toxicity, and the ease of product purification when using the former. 39 H H CH2I 2 SmI2, THF Me OH H H H I DMPU Me 72 % OH H 146 145 Scheme 55 A very similar methodology has been used to synthesise the 5,7 ring system and the strained cyclopropane moiety of the aromadendrane family of sesquiterpenoids (Scheme 56). 62 Again, the driving force for the reaction is the cleavage of the cyclobutane C-C bond of the [2+2] photoadduct. H CH2I SmI2, THF H H DMPU, rt, 51% CO2Et H 147 H CO2Et CO2Et 149 Major product 148 + H H EtO2C 150 Scheme 56 H CO2Et 151 40 After fragmentation, the radical is trapped by a pendant α,β-unsaturated ester moiety, leading to the formation of a cyclopropane ring. The stabilised radical (or anion?) intermediate allows this apparently contra-thermodynamic process to take place. Although the cyclopropane was the major product isolated, a small amount of an isomeric mixture of α,β-unsaturated compounds was formed by cyclisation followed by opening on either side of the cyclopropyl carbinyl system. The examples discussed above describe the synthesis of a number of terpenoids in which the critical step is the fragmentation of the “internal” cyclobutane bond. The cleavage of the “external” cyclobutane bond, however, allows the formation of a key intermediate in the preparation of the sesquiterpenoid trichodiene (Scheme 57). 63 In the critical step of the synthesis, SmI2 facilitated the desired cleavage of the external cyclobutane bond, to give the fragmented product in 95% yield. This fragmentation not only formed the cyclopentylcyclohexane system needed, but also introduced the exocyclic methylene group, which is a structural feature found in the natural product. SmI2, THF DMPU I 152 O 95% O 153 154 Trichodiene Scheme 57 A number of physiologically active alkaloids contain a spirocyclic skeleton. This moiety is easily accessed by a novel cyclobutane ring cleavage (Scheme 58). 64 The 41 regioselective ring opening was effected after treatment of the tricyclic precursor with SmI2 in THF-DMPU, to give the desired spirocyclic ketone in 68% yield. The unique biological activity (e.g. compounds of this structure have been used in probing the mechanisms involved in transsynaptic transmission of neuromuscular impulses) of these alkaloids has stimulated considerable interest in their synthesis. O O H H R N Boc 155 SmI2, THF R DMPU 68% R = α/β-n-C5H11 N Boc 156 Scheme 58 1.5.2.2 Cyclopropane containing substrates Alkyl radical cyclisations and tandem cyclisations are powerful aspects of the synthetic chemist’s arsenal. The corresponding ring-opening fragmentation reactions, which in most cases are disfavoured from both a kinetic and thermodynamic viewpoint, are, however, scarcer. An exception to this rule of thumb is the cyclopropylcarbinylhomoallyl radical rearrangement, which is both fast and thermodynamically favoured as a direct consequence of the cyclopropyl ring strain, as is neatly shown by the work of Motherwell. 65 The beauty of the reaction is that ring opening occurs under stereoelectronic control, leading to the fragmentation of the exocyclic C-C bond. The regio- and stereochemistry of the intermediate is thus determined by the initially constructed cyclopropyl ketone. 42 If no radical trap is incorporated into the molecule or added to the reaction mixture, only the fragmented methyl substituted derivative is formed. If a radical acceptor is present, the radical cascade reaction will dominate and allows entry into either spiroketones (Scheme 59) or fused bicyclic systems depending on the connective placement of the radical accepting chain. The reaction proceeds with alkenes and alkynes, and activating electron-withdrawing groups enhance the yield of the reaction. O i) SmI2, THF DMPU OAc ii) AcCl 157 57 % 158 Scheme 59 The reaction sequence, mechanism and intermediates allow not only tandem radical cyclisation reactions, but also allow capitalisation of the enolate anion chemistry through trapping of the intermediate samarium enolates with electrophiles in cases where carbonyl-type alkene activating groups are present. Attempted trapping with allyl bromide gave the allylated product in 37% yield and the epimeric ketone products of simple ring fragmentation in a combined 9% yield (Scheme 60). These experiments showed that it is possible to trap the Sm enolates with allyl bromide after a radical reaction sequence had taken place, in somewhat diminished yields. 43 O O O i) SmI2, THF DMPU + ii) allyl bromide 159 160 161 37% 9% Scheme 60 Although other reagents such as Bu3SnH or sodium naphthalenide can be used for this type of radical chemistry, the SmI2/DMPU system was by far the most useful in terms of avoiding the problems of reagent basicity and/or second electron transfer associated with these reducing agents. Subsequent to his first example of a simple cyclopropane ring cleavage (Scheme 61),52 Molander has further developed the cyclopropyl ring cleavage reaction by taking advantage of the reducing strength of SmI2 for further transformations. 66 O O 2 SmI2, cat. Fe(DBM)3 Me THF, 69% 162 Me 163 Me Scheme 61 The initially formed methylene radical can be further reduced to the corresponding carbanion and be trapped intramolecularly by a number of electrophiles (Scheme 62). These include ketones, esters, epoxides and aldehydes and lead to the formation of a variety of functionalised spirocyclic, bicyclic and tricyclic ring systems. 44 O O 2.5 SmI2 Me n m 8 HMPA OH n m O 164 Me 165a: n=0, m=1 61% 165b: n=1, m=1 79% 165c: n=1, m=2 30% Scheme 62 The reductive ring opening of α-cyclopropyl ketones with SmI2 has been used in the key step for the preparation of angular and linearly fused triquinanes (Scheme 63 and Scheme 64). The ring cleavages were effected by a SmI2-THF-MeOH system in good yield. 67 H H OMEM O OMEM SmI2, THF, 25°C MeOH, 86 % O H H 167 166 Scheme 63 H SmI2, -78°C to 25°C BzO O H BzO THF, MeOH, 65% O H OBz OBz 168 169 Scheme 64 The samarium(II) iodide promoted ring opening of cyclopropylogous α-hydroxy carbonyl compounds has been investigated as a possible strategy in the ongoing search for new 45 syntheses of the tricyclo[5.3.1.01,7]undecane system of taxanes. 68 Two types of substrates were used in the study: a range of cis-substituted cyclopropanes (Scheme 65) and compounds containing a bicyclo[3.1.0]system ring (Scheme 66). The reactions with substrates containing aldehyde moieties were carried out in THF at room temperature in the absence of HMPA, while for reactions with ketones, the addition of 8-10 equivalents of HMPA was necessary. The former set of substrates formed a range of fragmentation products including homoallylic ketones, δ-hydroxy ketones and β-methyl-γ-hydroxy ketones in various amounts depending on the R group. The mechanism of the reaction was proposed to proceed via the ketyl radical. This radical then undergoes one of two different rearrangements depending on which C-C bond of the cyclopropane ring cleaves. Further reduction and, in one case, a sequential β-elimination afforded the three different fragmentation compounds. O OH R rt 170 O O SmI2, HMPA + R 171 O R R = alkyl OH 172 + Me OH R 173 Scheme 65 The results of the reactions of the bicyclic compounds with SmI2 were of greater interest. The regioselectivity of the reaction depended on the nature of the carbonyl group present: aldehydes underwent an endo bond cleavage followed by a β-elimination of the hydroxy group to give cyclohexenes, whereas ketones formed their respective cyclopentanols resulting from simple exo C-C bond fragmentation (Scheme 66). The rationalisation of 46 these findings relied either on a steric repulsion argument or one based on Frontier Molecular Orbital theory. OH OH SmI2 SmI2 R (R = alkyl) R (R = H) H O O O 174 175 176 Scheme 66 The reductions of both α-haloketones to the corresponding enolates and the ring opening of α-cyclopropyl ketones have both been investigated independently. Beerli et al. have studied the effect of having both these functionalities present under reducing conditions (Scheme 67 and Scheme 68). 69 Although a ring opening and elimination sequence took place with a variety of reducing agents, only SmI2 and NaHTe gave good stereochemical control. With systems such as Li/NH3, Zn/AcOH, Cr(III), and Bu3SnH/AIBN, mixtures of the subsequent keto-alkene isomers were obtained, with the thermodynamically favoured trans product dominating. The fact that the reduction with SmI2 is highly stereoselective (selective production of the cis or trans product, depending on the stereochemistry at the halogenated carbon atom), in contrast with other reducing agents like Bu3SnH, led the authors to suggest a concerted reaction pathway, which would be similar to a Grob fragmentation. 70 Whether the reaction starts at the halogen or at the ketone is unclear, and it is possible that the mechanism resembles that proposed earlier for the ring scission of γ-haloesters (see section 3.1).38 47 Br H O O O SmI2, THF H + MeOH, rt H H H 178 179 99 1 76 % 177 Scheme 67 O Br H O O H SmI2, THF H MeOH, rt 85 % + 180 H H 181 98 182 2 Scheme 68 The reduction and subsequent ring opening of α-halo-oxirane rings has been well studied. These reactions are generally selective for carbon-oxygen bond cleavage, although carbon-carbon bond fragmentation does occur when the oxirane ring possesses a vinyl or aryl substituent, which would lead to a resonance stabilised carbon radical (Scheme 69). 71 Treatment of a bromomethyl epoxide with 2.2 equivalents of SmI2/THF in the presence of HMPA and MeOH at ambient temperature afforded the analogous allyl alcohol (1-phenyl-2-(1-naphthylmethoxy)ethane) as well as the vinyl ether (1-(1naphthyl)-3-phenylprop-2-enol) as a by-product. Br OH SmI2, HMPA Ph O Nap 183 MeOH, THF (Nap: 1-naphthyl) Ph Nap 184 Scheme 69 + Ph O 185 Nap 48 The presence of the vinyl ether was indicative of a radical fragmentation reaction. The ratio of the fragmentation products was dependent upon a number of factors. These included the ratio of HMPA to SmI2, the amount of methanol used, the concentration of the reaction mixture i.e. THF volume, the number of equivalents of SmI2 added and the reaction temperature. Dilution of the mixture favoured the C-C bond cleavage, as did an increase in reaction temperature; at -78 °C, no C-C fragmentation product was detected and 94% of the alcohol was recovered, while the highest percentage of C-C fragmentation took place at 50 °C. The total product yield did, however, suffer at elevated temperatures. The authors suggested that the reaction is initiated by reduction of the carbon-bromine bond to produce the oxiranylmethyl radical. Two pathways then become available. The first is a further reduction by a second equivalent of SmI2, followed by C-O cleavage to afford the Sm-alkoxide. The second possibility is a C-C radical fragmentation to give the vinyl ether radical intermediate. Further reduction yields the anion which either protonates to give the vinyl ether or it can recyclise and undergo C-O cleavage. The timing of the anionic quenching is critical in determining the product distribution. This is well illustrated in the graph shown below (Figure 3). If a proton source is present in the solution it is possible to rapidly trap the carbanion formed after reduction of the CC fragmentation intermediate (radical) to yield the vinyl ether before it can recyclise and eliminate oxygen. rel C-C frag. % 49 45 40 35 30 25 20 15 10 5 0 0 50 100 150 200 250 300 MeOH / equiv Figure 3. Effects of added MeOH on the relative ratio of C-C fragmentation. The effect of added HMPA is difficult to rationalise. Intuitively one would think that the increase in reducing potential with increasing HMPA concentration would favour the reduction of the initially formed oxiranylmethyl radical, and yield an increasing amount of C-O cleaved product. This is opposite to experimental facts. The relative C-C fragmentation product increases with increasing HMPA concentration up to a maximum at 8 equivalents of HMPA (Figure 4). Although SmI2 with 4-5 equivalents of HMPA is considered to be most effective for the reduction of primary alkyl radicals, the initial radical formed in this reaction is benzylic and hence delocalised. The authors tentatively rationalised the observation by concluding that the reduction of the carbon radical possessing bulky aryl substituents may be sensitive to steric factors if the process involves the formation of a carbon-Sm bond similar to that of primary alkyl radicals. As the amount of HMPA increases so does the size of the co-ordination complex, and thus the reduction of the initial radical would slow 50 down. This would allow the C-C fragmentation pathway to compete with the further rel C-C frag. % reduction.. 40 35 30 25 20 15 10 5 0 0 5 10 15 20 HMPA / equiv Figure 4. Effects of added HMPA on the relative ratio of C-C fragmentation SmI2 is able to promote the conversion of α-bromomethyl cyclic β-keto esters to the corresponding ring-expanded one-carbon homologated γ-ketoesters in good yields. 72 The approach involves an intramolecular samarium Barbier reaction followed by a ring expansion sequence. The mechanism proposed is depicted below (Scheme 70). Deuterium labelling experiments showed a large amount of deuterium incorporation in the ring-expanded product. This lends credence to the reaction pathway that involves a reduction of the radical intermediate, rather than hydrogen abstraction from the solvent. With R = Me, the cyclopropanol was recovered in 98% yield. The presence of the ester moiety is, therefore, crucial for the ring expansion that leads to a carbanion intermediate. When the ring size of the substrate was reduced to the cyclopentane derivative, the 51 addition of HMPA and a proton source were necessary for the ring expansion. In their absence, a naphthalene derivative was formed. O R SmI2 -Br- H R 187 SmI2 I2SmO R H 189 O 188 R SmI2 190 H+ HO O R Me R O 186 O O Br OSmI2 H+ Me OEt 191 192 193 Scheme 70 The literature on bicyclo[n.1.0] radicals reveals a preference for stereocontrolled exocyclic radical ring opening as opposed to the thermodynamically favoured endocyclic ring opening. 73 Exocyclic ring opening has been achieved utilising a variety of electron transfer techniques, including reagents such as SmI2. The selectivity of the reaction can be altered to favour the endocyclic C-C fragmentation reaction if an appropriately situated radical/anion stabilizing group, such as an ester moiety, is incorporated into the substrate (Scheme 71). 74 Thus, when a solution of the cyclopropyl compound (below) is treated with SmI2 in THF, the ring-expanded product is isolated in 44% yield. It was found that HMPA and DMPU were ineffective as additives in increasing the yield. However, when a proton source such as methanol was added, the reaction proceeded 52 smoothly and the yield increased two-fold. This work contrasts that of the exocyclic ring opening, in which no stabilizing group is present on the substrate molecule. O O SmI2 n CO2Bn 194 THF, MeOH 89% n = 1, 2 n CO Bn 2 195 Scheme 71 A number of natural products and key intermediates have been elegantly synthesised using SmI2 radical cascade methodology. (±)-Paeonilactone B has been constructed utilising such an approach (Scheme 72). 75 The mechanism presumably involves an intitial cyclisation of the ketyl radical onto a methylenecyclopropane unit with subsequent ‘endo’ ring opening to give the methylene cyclohexyl radical. This then cyclises onto the pendant alkyne giving rise to a cis-fused bicyclic system. The authors found that the addition of HMPA or DMPU was imperative for a successful reaction, with HMPA far out performing DMPU with respect to both yield and diastereoselectivity. The observed diasteroselectivity (cis-hydroxy vs trans-hydroxy, 10:1) is attributed to steric constraints in the transition state. The cyclisation is thought to proceed via a chair-like transition state, favouring the conformation in which both the bulky Sm(HMPA) enolate and the prop-2-ynyl ether adopt pseudo-equatorial positions. 53 O HO HO SmI2, t-BuOH, HMPA O H + THF, 0 °C O 196 199 O 200 HO I2SmO O I2SmO O O O O (±)-Paeonilactone B 197 198 201 Scheme 72 A similar methodology was applied to the corresponding allyl ethers in the hope of synthesizing paeonilactone A. When the keto-diene substrate was subjected to SmI2 / HMPA the fragmentation reaction proceeded as before, but the yield as well as the stereoselectivity at the newly formed chiral centre were reduced with respect to the analogous propargyl ether (Scheme 73). Subjecting the diastereomer to the same system, however, yielded a single diasteomeric bicyclic product (17% yield) accompanied by its dimer as a single diastereomer in 25% yield (Scheme 74). Although this particular reaction sequence was highly diastereoselective, it did not give the correct stereochemistry for the desired natural product. As noted before, the use of DMPU as a substitute for HMPA led to a loss of diastereoselectivity in the cyclisation step. Although the cyclisations of the allyl ethers failed to provide the correct stereochemistry for paeonilactone A, the conversion of the bicyclic ethers formed to diastereomers of the natural product were investigated. This allowed for the efficient and stereoselective synthesis of (±)-6-epi-paeonilactone A. 54 O SmI2, t-BuOH, HMPA O H HO HO THF, 0 °C H H + H H O 1:1 (35%) O 203 202 204 Scheme 73 O SmI2, ButOH, HMPA O H 205 THF, 0 °C OH HO HO H H H H + H O 206 17% O O H 207 25% Scheme 74 Walborski and Topolski 76 studied the reaction of chiral cyclopropyl halides with SmI2 in the presence of HMPA. The major product was the racemic reduced cyclopropyl compound, with the alkene and dimeric product being only minor components (Scheme 75). By carrying out these reactions in the presence of deuterated methanol, the authors were able to deduce which products were formed via a radical process and which were formed via an anionic sequence. Their experiments showed only a 15 % deuterium incorporation into the cyclopropyl derivative, implying that ring opening or H-atom abstraction from the solvent occurs before the radical can be further reduced. The alkene, however, possessed one deuterium atom per molecule, proving that it was formed after the radical had been reduced by a second equivalent of SmI2. 55 Ph Ph CH3 MeOD CH3 Ph [SmI2]+ 210 Ph 211 S-H SmI2 Ph Ph CH3 Br Ph CH3 Ph (S)-(+) 209 208 Ph CH3 SmI2 Ph CH2D MeOD CH3 Ph Ph 213 CH3 Ph CH3 Ph CH2 CH2 212 Ph Ph 214 Scheme 75 SmI2 can also induce the regioselective cleavage of phenylsulfonyl activated cyclopropyl ketones. 77 The cleavage of these cyclopropanes followed by β-elimination of phenylsulfonyl radical leading to β,γ-unsaturated ketones has been demonstrated (Scheme 76). Ph H H Ph PhO2S H Ph H O H Ph H H Ph SO2Ph + PhO2S H H SmI2, HMPA, tBuOH 215 PhO2S H O H 216 O Ph + Ph SO2Ph H O Ph + Ph Ph O 217 67% 218 9% Scheme 76 219 3% 56 The SmI2-Fe(DBM)3 [tris(dibenzoylmethido)iron(III)] reagent system has been used to successfully promote the ring opening reaction of a number of cyclopropane-1,1dicarboxylic esters. 78 Excellent yields were achieved in a relatively short period of time with a number of different ester moieties. When the same reactions were carried out at reflux temperature in the presence of an aliphatic ketone, the respective 5-pentanolide derivatives were isolated (Scheme 77). The addition of aldehydes or aromatic ketones resulted in a significant amount of pinacol products with low yields of the desired 5pentanolides. S,S'-Diphenyl cyclopropane-1,1-dicarbothioate was allowed to react with carbonyl compounds under similar conditions, giving δ-hydroxy esters in moderate yield. R1 CO2Me R H + R1COR2 SmI2, Fe(DBM)3 (4 mol%) CO2Me O CO2Me R THF, 220 O R2 222 221 Scheme 77 Yamashita and co-workers have expanded on the ring opening of cyclopropanecarboxylic esters and cyclopropane-1,1-dicarboxylic esters with a SmI2-HMPA-THF system in the presence of t-BuOH as a proton source to give 4-substituted butyric esters and (2substituted ethyl)malonic esters (Scheme 78). 79 H CO2Et H CO2Et R SmI2 THF, 223 R R H COOEt COOEt COOEt COOEt 224 Scheme 78 57 In the absence of a proton source, however, they were able to reductively dimerise several 2-substituted cyclopropane-1,1-dicarboxylic esters. 80 The initial yields were poor, but were increased by the omission of HMPA from the system. The yield was further improved by conducting the reactions in a solution of SmI2-THF under reflux. 1.5.3 1,4-Diketones in non-strained and strained systems Hoffmann and co-workers 81 have recently described a 1,4-pinacolisation methodology utilising SmI2 that is useful for the production of highly strained systems containing a 1,2-cyclobutanediol moiety. Ghosh anticipated that this methodology would be useful for gaining easy access to [3.3.2]propellanes. Thus, bicyclo[2.2.1]heptane derivatives bearing a 1,4-dicarbonyl moiety were prepared and subjected to reaction with SmI2. 82 Contrary to expectations of cyclisation of the substrate, fragmentation to macrocyclic compounds was observed (Scheme 79). Although the pinacol reaction is a possible reaction outcome, it would lead to an increase in strain of an already strained norbornene system: the more facile C-C fragmentation pathway is, therefore, followed. Relieving some ring strain by prior reduction of the double bond did not alter the outcome of the reaction. The addition of HMPA might have been a deciding factor in this reaction. MeOC H SmI2, THF COMe HMPA, t-BuOH COMe 100% 225 H MeOC 226 Scheme 79 58 Camps has recently reported a similar experience of the fragmentation of a 1,4-diketone while trying the Hoffmann pinacol methodology. 83 Upon subjecting the strained bisnoradamantane system to SmI2, it readily underwent fragmentation to give a mixture of three stereoisomeric bicyclic diketones in 80% yield (Scheme 80). A mixture of stereoisomeric alcohols (16% yield) derived from further reduction of the diketones was also isolated. Me Me t-Bu-OC Me CO-t-Bu t-Bu HO SmI2, THF 227 Me t-Bu OH 228 Me t-Bu-OC CO-t-Bu + diastereomers Me 230 229 Me + t-Bu-OC Me H C OH t-Bu 231 Scheme 80 A molecular mechanics (MM2 and MM3) investigation was carried out on the substrate, the possible cyclobutanediol isomers, and the fragmented diketones giving the formation enthalpies as well as the strain energies of the individual molecules. The calculations confirmed natural intuition, showing an enormous increase in strain energy for the pinacol products, while the strain energy of the products after fragmentation was considerably reduced. The authors went further and stated that it was reasonable to 59 assume that the transition-state for the conversion of the diketyl radical derived from the bisnoradamantane diketone to the bis-enolate derived from the fragmented product is of much lower energy than the corresponding transition-state for its conversion to the diolate derived from the pinacol product. The fragmentation pathway via the diketyl radical is therefore more facile than that of its pinacol counterpart. 1.6 Three and Four Membered Rings One of the ways in which SmI2 is made is the reaction of samarium metal with diiodomethane. The intermediate formed is ‘ISmCH2I’ which undergoes α-elimination generating SmI2 and methylene. This enables one to trap the carbenoid intermediate with olefins thus producing cyclopropanes and providing a useful alternative to the traditional Simmons-Smith procedure. In practice, a more economical reagent to use for the cyclopropanation is the Sm(Hg) amalgam. The SmI2-mediated reaction offers enhanced chemoselectivity and often higher diastereoselectivity than in classical Simmons-Smith reactions (Scheme 81). 84 CF3 OH Sm(Hg), CH2I2 OH C6H13 THF 232 CF3 H C6H13 H 233 Scheme 81 Enolates are inert to cyclopropanation with the Simmons-Smith reagent system and form α-methylated ketones instead. By contrast generation of the kinetic enolate with LDA 60 followed by the addition of SmI2/CH2I2 generates cyclopropanes in good yield (Scheme 82). 85 O OH 1. LDA, THF 2. SmI2, CH2I2 234 235 Scheme 82 The methodology can be extended to α-halo ketones. The enolate is generated in situ by reaction with SmI2 and cyclopropanation follows (Scheme 83). 86 O HO 3 CH2I2, 2 SmI2 Br THF 236 237 Scheme 83 It is possible to directly convert carboxylic acid esters to cyclopropanols by reacting them with diiodomethane and SmI2 (generated in situ). The reaction proceeds via a nucleophilic acyl substitution reaction forming the corresponding iodomethyl ketones. Reduction of this α-hetrosubstituted ketone by SmI2 generates the enolate, which is finally trapped with the carbenoid intermediate to give the cyclopropane product (Scheme 84).85 O R O CH2I2 OR' 238 Sm OSmI2 SmI2 R CH2I 239 R CH2 240 Scheme 84 CH2I2 OH R Sm 241 61 The samarium Barbier reaction of nonracemic β-chloro-substituted amides provides high yields of the corresponding cyclopropanols with good diastereoselectivity. 87 The Sm(III) by-product acts an effective Lewis acid and controls the stereoselectivity of the reaction by chelating the functional groups (Scheme 85). O O O O 2 SmI2 Cl N THF, HMPA O HO N CH2Ph CH2Ph 242 243 Scheme 85 One of the more popular applications of the Barbier reaction is the synthesis of bicyclic alcohols (Scheme 86). The construction of cyclopropanols is accomplished by treating αtosyloxymethyl cyclohexanones with SmI2. 88 O OTs HO 2 SmI2 THF 244 245 Scheme 86 A similar reaction was attempted to produce a cyclobutanol derivative but the cyclisation did not occur. Rather, a reductive β-elimination took place generating a samarium enolate (Scheme 87).87 62 I O O OSmI2 SmI2 THF 246 248 247 Scheme 87 The formation of cyclopropanols in exceedingly good yields from β-bromo ketones or aldehydes is particularly impressive. 89 The substrate is usually prepared in situ from 3bromopropionates via a Grignard reaction. The analogous reaction to produce cyclobutanes has also been attempted but much poorer yields were obtained (Scheme 88). Br OEt 1. RMgX n O 249 n 2. SmI2, THF-HMPA R OH 250 n = 1 Yield = 70-99% =2 = 5% Scheme 88 Nucleophilic acyl substitution reactions with esters are, however, amenable to the synthesis of four membered rings. The reactions are accomplished under mild conditions upon treatment with SmI2 (Scheme 89). 90 I CO2Et Ph O 2 SmI2 THF, cat. Fe(III) 74% Ph 252 251 Scheme 89 63 Cyclopropanols are readily accessible via a sequential process involving an acyl radical cyclisation followed by an intermolecular Barbier reaction. 91 Thus 2-allyloxybenzoyl chloride reacts with SmI2 generating an acyl radical. This undergoes intramolecular addition to the double bond, thereby forming a new radical. The radical is further reduced by a second equivalent of SmI2 to form the organosamarium intermediate, which reacts with the ketone giving the desired cyclopropanol (Scheme 90). O HO 2 SmI2 Cl THF O 253 O 254 Scheme 90 In a similar vein, sequential processes have been developed to allow access to bicyclic structures containing the cyclobutyl moiety. 92 These protocols are also commonly used in the synthesis of heterocyclic systems (Scheme 91). Both activated alkynes and unactivated olefins can be used as radical traps in the reaction. O I EtO O SmI2 SiMe3 THF-HMPA 255 OH O 256 Scheme 91 SiMe3 64 Another intramolecular Barbier reaction allows access to cyclobutanols with an alkenyl moiety. The same protocol was used as in the previous example except that the substrate is set up for β-elimination after the bicyclisation has occurred (Scheme 92). 93 HO O EtO O Br SmI2 O O HO THF-HMPA 259 258 257 Scheme 92 Another way one can envisage making small ring systems is via radical cyclisation of a suitable intermediate onto the C-C double bond of α,β-unsaturated esters. Guibé and co-workers have shown that δ-iodo and δ-bromo-α,β-unsaturated esters with various substituents at the β- and γ-positions readily cyclise to form cyclopropane compounds under the reducing power of SmI2 and in the presence of a proton source (Scheme 93). 94 CO2Bn I SmI2, THF CO2Bn t-BuOH 261 260 Scheme 93 The attempted SmI2-mediated cyclisation reaction of a δ-iodo-α,β-unsaturated ester to afford a cyclobutanol failed, giving the reduced product in its stead (Scheme 94). 95 65 O O OEt 1.5 SmI2, cat. NiI2 OEt THF, t-BuOH I 263 262 Scheme 94 In order to obtain the desired cyclobutanol product in these types of reactions it is essential to make use of the gem-disubstitution effect. 96 This was well illustrated by Weinges and co-workers who successfully cyclised a gem-dimethyl-γ,δ-unsaturated aldehyde to the corresponding cyclobutanol in the presence of SmI2 (Scheme 95). 97 This work was ellaborated on by Procter and co-workers who managed to improve the reaction conditions and yields but were unable to obtain any cyclobutanol product without some sort of disubstitution in the substrate. 98 BnO CHO SmI2 CO2Et BnO OH THF-HMPA 264 CO2Et 265 Scheme 95 Yet another method of synthesising cyclobutane derivatives is that of the pinacol coupling reaction of 1,4-diketone substrates mediated by SmI2. This methodology, developed by Hoffmann and co-workers, allows for easy access into a variety of substituted 1,2-cyclobutanediol derivatives. 99 The beauty of the reaction stems from the 66 fact that no gem-disubstitution is necessary and the reaction requires no co-solvents or additives (Scheme 96). O R2 R1 O HO SmI2 OH R1 THF 266 R2 267 Scheme 96 The pinacol reaction is also very powerful and enables the formation of extremely ring strained compounds (Scheme 97). 100 O R R SmI2 H THF OH OH O 269 268 Scheme 97 1.7 Conclusions 1.7.1 Fragmentations SmI2, either in the presence or absence of co-solvents, promotes manifold reactions, many of which are fragmentation reactions. In a number of cases, the fragmentation methodology has been put to good use in the preparation of highly functionalised products, which often are natural products or those that possess physiological activity. The fragmentation protocol has been found useful in a variety of transformations, and has 67 been effectively applied to sequential reactions in which one or more transformations follow the initial fragmentation step. SmI2 has been found to be the reductant of choice for many reductive fragmentation reactions, and generally affords yields higher than those provided by other reducing agents, and in most instances affords higher chemical and stereochemical yields. This being the case, SmI2 should continue to find application in many synthetic sequences in more and more laboratories engaged in organic synthesis. 1.7.2 Small Ring Systems Samarium diiodide has been used to synthesise a number of small ring systems with good stereoselectivity. The use of carbohydrates as precursors to these compounds and hence the production of valuable, chiral small ring systems is, however, lacking. The methodologies presented in the literature have also not been tested on complex molecules such as sugars where the potential for fragmentation reactions is elevated. The current synthetic routes to three and four membered ring systems lack the capacity to produce highly substituted end products. These areas require attention and will be addressed in this manuscript. 1.8 References 1 (a) Girard, P., Namy, J.L., Kagan, H.B. J. Am. Chem. Soc. 1980, 102, 2693. (b) Molander, G.A. Chem. Rev. 1992, 92, 29. (c) Sasaki, M., Collin, J., Kagan, H.B. New J. Chem. 1992, 16, 89. 68 2 (a) Inanaga, J., Ishikawa, M., Yamaguchi, M. Chem. Lett. 1987, 1485. (b) Ruder, S.M. Tetrahedron Lett. 1992, 33, 2621. (c) Cabri, W., Candiani, I., Colombo, M., Franzoi, L., Bedeschi, A. Tetrahedron Lett. 1995, 36, 949. (d) Hasegawa, E., Curran, D.P. J. Org. Chem. 1993, 58, 5008. 3 (a) Ogawa, A., Takami, N., Sekiguchi, M., Ryu, I., Kambe, N., Sonoda, N. J. Am. Chem. Soc. 1992, 114, 8729. (b) Ogawa, A., Nanke, T., Takami, N., Sumino, Y., Ryu, I., Sonoda, N. Chem. Lett. 1994, 379. 4 Lu, X., Ma, S., Zhu, J. Tetrahedron Lett. 1988, 29, 5129. 5 Bennet, S.M., Larouche, D. Synlett 1991, 805. 6 (a) Fevig, T.L., Elliot, R.L., Curran, D.P. J. Am. Chem. Soc. 1988, 110, 5064. (b) Molander, G.A., Kenny, C. J. Am. Chem. Soc. 1989, 111, 8236. (c) Molander, G.A., Harring, L.S. J. Org. Chem. 1990, 55, 6171. (d) Inanaga, J., Ujikawa, O., Yamaguchi, M. Tetrahedron Lett. 1991, 32, 1737. 7 Fukuzawa, S.,Tsuchimoto, T. Synlett 1993, 803. 8 Aurrecoechea, J.M., Fernandez-Acebes, A. Tetrahedron Lett. 1993, 34, 549. 9 Batey, R.A., Motherwell, W.B. Tetrahedron Lett. 1991, 32, 6649. 10 (a) Sturino, C.F., Fallis, A.G. J. Am. Chem. Soc. 1994, 116, 7447. (b) Sturino, C.F., Fallis, A.G. J. Org. Chem. 1994, 59, 6514 11 (a) Fukuzawa, S, Nakanishi, A., Fujinami, T., Sakai, S. J. Chem. Soc. Chem. Commun. 1986, 624. (b) Fukuzawa, S, Nakanishi, A., Fujinami, T., Sakai, S. J. Chem. Soc. Perkin Trans. 1 1988, 1669. (c) Otsubo, K., Inanaga, J., Yamaguchi, M. Tetrahedron Lett. 1986, 27, 5763. 12 Ujikawa, O., Inanaga, J., Yamaguchi, M. Tetrahedron Lett. 1989, 30, 2837. 69 13 Inanaga, J., Katsuki, J., Ujikawa, O., Yamaguchi, M. Tetrahedron Lett. 1991, 32, 4921. 14 Kito, M., Sakai, T., Yamada, K., Matsuda, F., Shirahama, H. Synlett 1993, 158. 15 Baldwin, J.E., Turner, S.C.M., Moloney, M.G. Tetrahedron 1994, 50, 9411. 16 Kan, T., Nara, S., Ito, S., Matsuda, F., Shirahama, H. J. Org. Chem. 1994, 59, 5111. 17 Shim, S.C., Hwang, J.-T., Kang, H.-Y., Chang, M.H. Tetrahedron Lett. 1990, 31, 4765. 18 Molander, G.A., McKie, J.A. J. Org. Chem. 1995, 60, 872. 19 Molander, G.A., McKie, J.A. J. Org. Chem. 1992, 57, 3132. 20 Bannai, K., Tomimori, K., Kato, Y., Kurozumi, S., Nyori, R. Tetrahedron 1990, 46, 6689. 21 Gillmann, T. Tetrahedron Lett. 1993, 34, 607. 22 (a) Shiue, J.-S., Lin, C.-C., Fang, J.-M. Tetrahedron Lett. 1993, 34, 335. (b) Kise, N., Suzumoto, T., Shono, J. J. Org. Chem. 1994, 59, 1407. 23 (a) Namy, J.L., Souppe, J., Kagan, H.B. Tetrahedron Lett. 1983, 24, 765. (b) Fürstner, A., Csuk, R., Rohrer, C., Weidmann, H.J. J. Chem. Soc., Perkin Trans. 1 1988, 1729. (c) Akane, N., Kanagawa, Y., Nishiyama, Y., Ishii, Y. Chem. Lett. 1992, 2431. 24 (a) Enholm, E.J., Forbes, D.C., Holub, D.P. Synthetic Commun. 1990, 20, 981. (b) Imamoto, T., Nishimura, S. Chem. Lett. 1990, 1141. 25 (a) Chiara, J.L., Cabri, W., Hanessian, S. Tetrahedron Lett. 1991, 32, 1125. (b) Chiara, J.L., Martin-Lomas, M. Tetrahedron Lett. 1994, 35, 2969. (c) Uenishi, J., Masuda, S., Wakabayashi, S. Tetrahedron Lett. 1991, 32, 5097. 26 Molander, G.A., Kenny, C.J. J. Org. Chem. 1988, 53, 2132. 27 Clayden, J., Julia, M. J. Chem. Soc., Chem. Commun. 1994, 2261. 70 28 Molander, G.A., Etter, J.B. Synth. Commun. 1987, 17, 901. 29 Molander, G.A., Etter, J.B., Zinke, P.W. J. Am. Chem. Soc., 1987, 109, 453. 30 Suginome, H., Yamada, S. Tetrahedron Lett. 1987, 28, 3963. 31 (a) Sosnowski, J.J., Danaher, E.B., Murray, R.K. Jr. J. Org. Chem. 1985, 50, 2759. (b) Molander, G.A., McKie, J.A. J. Org. Chem. 1991, 56, 4112. 32 Kagan, H.B., Namy, J.L., Girard, P. Tetrahedron 1981, 37, 175, Suppl. 1. 33 Zhang, Y., Liu, T., Lin, R. Synth. Commun. 1988, 18, 2003. 34 Arime, T., Kato, N., Komadate, F., Seegusa, H., Mori, N. Synthetic Commun. 1994, 24, 3315. 35 Molander, G.A., Etter, J.B., Harring, L.S., Thorel, P.-J. J. Am. Chem. Soc. 1991, 113, 8036. 36 Molander, G.A., McKie, J.A. J. Org. Chem. 1993, 58, 7216. 37 Fevig, T.L., Elliot, R.L., Curran, D.P. J. Am. Chem. Soc. 1988, 110, 5064. 38 Curran, D.P., Totleben, M.J. J. Am. Chem. Soc. 1992, 114, 6050. 39 Molander, G.A., Hahn, G. J. Org. Chem. 1986, 51, 1135. 40 Lannoye, G.,Cook, J.M. Tetrahedron Lett. 1988, 29, 171. 41 Molander, G.A. Chem. Rev. 1992, 92, 29. 42 (a) Hasegawa, E., Curran, D.P. J. Org. Chem. 1993, 58, 5008. (b) Curran, D.P., Wolin, R.L. Synlett. 1991, 317. 43 Hojo, M., Aihara, H., Hosomi, A. J. Am. Chem. Soc. 1996, 118, 3533. 44 Shabangi, M., Flowers, R.A., II Tetrahedron Lett. 1997, 38, 1137. 45 Hou, Z., Wakatsuki, Y. J. Chem. Soc. Chem. Commun. 1994, 1205. 46 Hasegawa, E., Curran, D.P. Tetrahedron Lett. 1993, 34, 1717. 71 47 (a) Fevig, T.L., Elliot, R.L., Curran, D.P. J. Am. Chem. Soc. 1988, 110, 5064. (b) Molander, G.A., Harring, L.S. J. Org. Chem. 1990, 55, 6171. (c) Shiue, J.-S., Lin, C.-C., Fang, J.-M. Tetrahedron Lett. 1993, 34, 335. 48 Hou, Z., Zhang, Y., Wakatsuki, Y. Bull. Chem. Soc. Jpn. 1997, 70, 149. 49 Evans, W.J., Gummersheimer, T.S., Ziller, J.W. J. Am. Chem. Soc. 1995, 117, 8999. 50 Shotwell, J.B., Sealy, J.M., Flowers, R.A. II J. Org. Chem. 1999, 64, 5251. 51 Ananthanarayan, T.P., Gallagher, T. Magnus, P. J. Chem. Soc., Chem. Commun. 1982, 709. 52 Honda, T., Naito, K., Yamane, S.-i., Suzuki, Y. J. Chem. Soc., Chem. Commun. 1992, 1218. 53 Molander, G.A., McKie, J.A. J. Org. Chem. 1991, 56, 4112. 54 (a) Honda, T., Yamane, S.-i., Naito, K., Suzuki, Y. Heterocycles 1994, 37, 515. (b) Honda, T., Ishikawa, F., Yamane, S.-i. J. Chem. Soc., Chem. Commun. 1994, 499. (c) Honda, T., Yamane, S.-I., Naito, K., Suzuki, Y. Heterocycles 1995, 40, 301. (d) Honda, T., Ishikawa, F., Yamane, S.-I. J. Chem. Soc., Perkin Trans. 1 1996, 1125. (e) Honda, T., Yamane, S.-I., Ishikawa, F., Katoh, M. Tetrahedron 1996, 52, 12177. (f) Honda, T., Katoh, M., Yamane, S.-I. J. Chem. Soc., Perkin Trans. 1 1996, 2291 . 55 56 Honda, T., Ishikawa, F. J. Org. Chem. 1999, 64, 5542. Molander, G.A., Etter, J.B., Zinke, P.W. J. Am. Chem. Soc 1987, 109, 453. 57 Kang, H.-Y., Sang Hong, W. Tetrahedron Lett. 1995, 42, 7661. 58 Meng, Q., Hesse, M. "Topics in Current Chemistry", 1992, Vol. 161, 107, Springer- Verlag. 59 Goulaouic-Dubois, C., Guggisberg, A., Hesse, A. Tetrahedron 1995, 51, 12035. 72 60 Lange, G.L., Gottardo, C. Tetrahedron Lett. 1994, 35, 6607. 61 Lange, G.L., Gottardo, C., Merica, A. J. Org. Chem. 1999, 64, 6738. 62 Lange, G.L., Merica, A. Tetrahedron Lett. 1999, 40, 7897. 63 Lange, G.L., Furlan, L., MacKinnon, M.C. Tetrahedron Lett. 1998, 39, 5489. 64 Comins, D.L., Zheng, X. J. Chem. Soc., Chem. Commun. 1994, 2681. 65 (a) Batey, R.A., Motherwell, W.B. Tetrahedron Lett. 1991, 32, 6211. (b) Batey, R.A., Harling, J.D., Motherwell, W.B. Tetrahedron 1996, 52, 11421. 66 Molander, G.A., Alonso-Alija, C. Tetrahedron 1997, 53, 8067. 67 Hwang, J.-T., Liao, C.-C. Tetrahedron Lett. 1991, 32, 6583. 68 (a) Nivlet, A., Le Guen, V., Dechoux, L., Le Gall, T., Mioskowski, C. Tetrahedron Lett. 1998, 39, 2115. (b) Nivlet, A., Dechoux, L., Le Gall, T., Mioskowski, C. Eur. J. Org. Chem. 1999, 3251. 69 Beerli, R., Brunner, E.J., Borschberg, H.-J. Tetrahedron Lett. 1992, 33, 6449. 70 Grob, A. Angew. Chem. Int. Ed. Engl. 1965, 8, 535. 71 Hasegawa, E., Takahashi, M., Horaguchi, T. Tetrahedron Lett. 1995, 36, 5215. 72 Hasegawa, E., Kitazume, T., Suzuki, K., Tosaka, E. Tetrahedron Lett. 1998, 39, 4059. 73 (a) Cristol, S., Barbour, R.V. J. Am. Chem. Soc. 1968, 90, 2832. (b) Beckwith, A.L.J., Philliou, G.J. J. Chem. Soc. Chem Commun. 1971, 658. (c) Beckwith, A.L.J., Philliou, G.J. Aust. J. Chem. 1976, 29, 123. (d) Friedrich, E.C., Holmstead, R.L. J. Org. Chem. 1971, 36, 971. (e) Ingold, K.U., Walton, J.C., Acc. Chem. Res. 1986, 19, 72. 74 Lee, P.H., Lee, J. Tetrahedron Lett. 1998, 39, 7889. 75 a) Boffey, R.J., Whittingham, W.G., Kilburn, J.D. J. Chem. Soc., Perkin Trans. 1 2001, 487. (b) Boffey, R.J., Santagostino, M., Whittingham, W.G., Kilburn, J.D. J. Chem. Soc., 73 Chem. Commun. 1998, 1875. (c) Bofey, R.J., Whittingham, W.G., Kilburn, J.D. Tetrahedron Lett. 1999, 40, 5625. 76 Walborsky, H.M., Topolski, M., J. Org. Chem. 1992, 57, 370. 77 Reutrakul, V., Saeeng, R., Pohmakotr, M., Kongsaeree, P. Tetrahedron Lett. 1999, 40, 1019. 78 Imamoto, T., Hatajima, T., Yoshizawa, T. Tetrahedron Lett. 1994, 35, 7805. 79 (a) Yamashita, M., Okuyama, K., Ohhara, T., Kawasaki, I., Ohto, S. Chem. Pharm. Bull. 1995, 43, 708. (b) Yamashita, M., Okuyama, K., Ohhara, T., Kawasaki,I., Sakai, K., Nakata, N., Kawabe, T., Kusumoto, M., Ohto, S. Chem. Pharm. Bull. 1995, 43, 2075. 80 Yamashita, M., Okuyama, K., Ohhara, T., Kawasaki, T., Ohta, S. Synlett. 1996, 547. 81 (a) Hoffmann, H.M.R., Munnich, I., Nowitzki, O., Stucke, H., Williams, D.J. Tetrahedron 1996, 52, 11783. (b)Nowitzki, O., Munnich, I., Stucke, H., Hoffmann, H.M.R. Tetrahedron 1996, 52, 11799. 82 Haque, A., Ghosh, S. J. Chem. Soc., Chem. Commun. 1997, 2039. 83 Camps, P., Lukach, A.E., Vázquez, S. Tetrahedron 2001, 57, 2419. 84 (a) Molander, G.A., Etter, J.B. J. Org. Chem. 1987, 52, 3942. (b) Molander, G.A., Harring, L.S. J. Org. Chem. 1989, 54, 3525. (c) Yamazaki, T., Lin, J.T., Takeda, M., Kitazume, T. Tetrahedron: Assymetry 1990, 1, 351. 85 Immamoto, T., Takiyama, N. Tetrahedron Lett. 1987, 28, 1307. 86 Immamoto, T., Hatajima, T., Takiyama, N., Takeyama, T., Kamiya, Y., Yoshizawa, T. J. Chem. Soc., Perkin Trans. 1 1991, 3127. 87 Fadel, A. Tetrahedron: Assymetry 1994, 5, 531. 88 Berks, A. Ph.D. Thesis, University of Colorado, Boulder, 1988. 74 89 Fukuzawa, S.-i., Furuya`, H., Tsuchimoto, T. Tetrahedron 1996, 52, 1953. 90 Molander, G.A., McKie, J.A. J. Org. Chem. 1993, 58, 7216. 91 Sasaki, M., Collin, J., Kagan, H.B. Tetrahedron Lett. 1988, 29, 6105. 92 Molander, G.A., Harris, C.R. Tetrahedron Lett. 1998, 54, 3321. 93 Molander, G.A., Harris, C.R. J. Org. Chem. 1998, 63, 4374. 94 David, H., Afonso, C., Bonin, M., Doisneau, G., Guillerez, M.-G., Guibé, F. Tetrahedron Lett. 1999, 8557. 95 Molander, G.A., Harris, C.R. J. Org. Chem. 1997, 62, 7418. 96 (a) Jung, M.E., Marquez, R., Houk, K.N. Tetrahedron lett. 1999, 40, 2661. (b) Park, S.- U., Varick, T.R., Newcomb, M. Tetrahedron Lett. 1990, 31, 2975. 97 Weinges, K., Schmidbauer, S.B., Schick, H. Chem. Ber. 1994, 127, 1305. 98 (a) Johnston, D., McCusker, C.M., Procter, D.J. Tetrahedron Lett. 1999, 4913. (b) ) Johnston, D., McCusker, C.F., Muir, K., Procter, D.J. J. Chem. Soc., Perkin Trans. 1 2000, 681. 99 Hoffmann, H.M.R., Munnich, I., Nowitzki, O., Stucke, H., Williams, D.J. Tetrahedron 1996, 52, 11783. 100 Nowitzki, O., Munnich, I., Stucke, H., Hoffmann, H.M.R. Tetrahedron 1996, 52, 11799.