An SRY-negative 47,XXY mother and daughter
advertisement

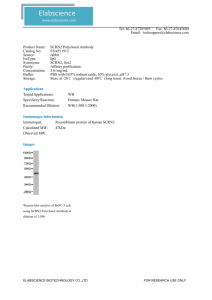
Cytogenet Cell Genet 91:204–207 (2000) An SRY-negative 47,XXY mother and daughter S. Röttger,a K. Schiebel,b G. Senger,c S. Ebner,c W. Schemppa and G. Scherera a Institut für Humangenetik und Anthropologie, Universität Freiburg, Freiburg; für Humangenetik, Universität Heidelberg, Heidelberg; and c Praxis für Medizinische Genetik, Regensburg (Germany) b Institut Dedicated to Professor Dr. Ulrich Wolf on the occasion of his retirement. Abstract. Females with XY gonadal dysgenesis are sterile, due to degeneration of the initially present ovaries into nonfunctional streak gonads. Some of these sex-reversal cases can be attributed to mutation or deletion of the SRY gene. We now describe an SRY-deleted 47,XXY female who has one son and two daughters, and one of her daughters has the same 47,XXY karyotype. PCR and FISH analysis revealed that the mother carries a structurally altered Y chromosome that most likely resulted from an aberrant X-Y interchange between the closely related genomic regions surrounding the gene pair PRKX and PRKY on Xp22.3 and Yp11.2, respectively. As a consequence, Yp material, including SRY, has been replaced by terminal Xp sequences up to the PRKX gene. The fertility of the XXY mother can be attributed to the presence of the additional X chromosome that is missing in XY gonadal dysgenesis females. To our knowledge, this is the first human XXY female described who is fertile. The chromosome constitution 47,XXY occurs in 1 in 1,000 conceptions but, due to spontaneous abortion, is seen in only 1 in 2,000 livebirths (Hook and Hamerton, 1977; Jacobs et al., 1988). Individuals with a 47,XXY karyotype are almost exclusively males, presenting with the clinical phenotype of Klinefelter syndrome (Jacobs and Strong, 1959). A few exceptional 47,XXY females have been described. Five of these rare cases have been diagnosed as suffering from androgen insensitivity (testicular feminization) syndrome resulting from mutations in the androgen receptor gene (German and Vesell, 1966; BartschSandhoff et al., 1976; Gerli et al., 1979; Müller et al., 1990; Uehara et al., 1999). One 47,XXY 6-yr-old girl had female external genitalia, a uterus, and testes in the labia majora (Schmid et al., 1992). Another 47,XXY female, 15 yr old, had essentially normal female external and internal genitalia and one gonad with ovarian stroma but no follicles, the other gonad containing tubule-like structures devoid of germ cells (Thangaraj et al., 1998). These seven 47,XXY females were all sterile and had normal Y chromosomes with (where investigated) a normal testis-determining SRY gene. The chromosome constitution 47,XXX is usually found only in females. Again, three exceptional 47,XXX males have been described, all three resulting from an aberrant X-Y interchange leading to transfer of Yp material, including SRY, to one of the X chromosomes (Annerén et al., 1987; Müller et al., 1987; Scherer et al., 1989). We now describe the mirror image of these 47,XXX males, a 47,XXY female resulting from an aberrant X-Y interchange with transfer of Xp material onto Yp, with concomitant loss of SRY. The same 47,XXY chromosome constitution was found in one of her daughters. Copyright © 2001 S. Karger AG, Basel Materials and methods S.R. and K.S. contributed equally to this work. K.S.’s present address is Institut für Biochemie, Universität Erlangen-Nürnberg, Erlangen (Germany). Received 18 August 2000; revision accepted 13 September 2000. Request reprints from Dr. Gerd Scherer, Institut für Humangenetik und Anthropologie der Universität Freiburg, Breisacher Strasse 33, D–79106 Freiburg (Germany); telephone: 49-761-270-7030; fax: 49-761-270-7041; e-mail: scherer@humangenetik.ukl.uni-freiburg.de ABC Fax + 41 61 306 12 34 E-mail karger@karger.ch www.karger.com © 2001 S. Karger AG, Basel 0301–0171/00/0914–0204$17.50/0 Cytogenetic and FISH analyses Chromosome analysis on amniotic fluid cells and peripheral blood lymphocytes was performed by GTG-banding using standard procedures. For fluorescence in situ hybridization (FISH) analysis, metaphase spreads were prepared by standard techniques from lymphoblastoid cells from the subject, R.M. Prior to FISH, the slides were treated with RNase, followed by pepsin digestion as described by Ried et al. (1992). Two-color FISH with humanderived DNA probes essentially followed the methods described in Schempp et al. (1995). The probes used for PRK, ARSE, and SHOX were, respectively, PAC 152F08 (Schiebel et al., 1997a; kindly provided by Gudrun Rappold, Accessible online at: www.karger.com/journals/ccg Heidelberg), cosmid cos61C4 (Franco et al., 1995; kindly provided by Kay Taylor, London), and cosmid LLNOYCO3)M)34F5 (Rao et al., 1997; kindly provided by Gudrun Rappold, Heidelberg). Fluorescence microscopy and imaging Preparations were evaluated using a Zeiss Axiophot epifluorescence microscope equipped with single-bandpass filters for excitation of red, green, and blue signals (Chroma Technologies). During exposures, only excitation filters were changed, allowing for pixel shift–free image recording. Images of high magnification and resolution were obtained using a black-and-white CCD camera (Photometrix Kodak KAF 1400) connected to the Axiophot microscope. Camera control and digital image acquisition involved the use of an Apple Macintosh Quadra 950 computer. PCR analyses PCR analyses for SRY and the Xp and Yp pseudoautosomal boundary were performed using primers and conditions as described previously (Jäger et al., 1990a, b). PCR analyses for PRKY and flanking sequences were performed with primer pairs for sY68, exon 3, exon 2, exon 1, TranD, and AMELY, as described earlier (Schiebel et al., 1997b). Y-specific primers TranY1for (5) TGT GCG GTC AGA CCC CAA CC 3)) and TranY1rev (5) CCT TCC TTC CTT CTT TCC TTC C 3)) were selected by comparison of Xspecific (GenBank accession number AC010104) and Y-specific (GenBank NT_003007) BAC sequences from GenBank. These primers are located 16 kb upstream of the first PRKY exon and revealed an amplification product of 368 bp (annealing temperature of 67 ° C). Results During the third pregnancy of R.M., who had previously given birth to a normal son and daughter, a routine prenatal diagnosis was performed at 16 wk of gestation because of her advanced maternal age of 33½ yr. A 47,XXY karyotype diagnostic for Klinefelter syndrome was found in all of the 16 investigated fetal amniocytes derived from two independent cell-culture flasks. At birth, however, the newborn presented with a female, not male, phenotype, with normal female external genitalia, and with bilateral clubbed feet. Karyotype analysis from peripheral blood lymphocytes of the newborn girl, B.M., confirmed the cytogenetic result obtained during pregnancy: 47,XXY, with no indication of mosaicism (15 metaphases analyzed). Additionally, the karyotypes of both parents were investigated. In the father, a normal 46,XY male karyotype was found, whereas cytogenetic analysis of 13 metaphases from peripheral lymphocytes of the mother, R.M., unexpectedly revealed the same 47,XXY karyotype as her daughter B.M. Again, no evidence for mosaicism was found, and the sex chromosomes and autosomes were of apparently normal structure. We assumed that the XXY sex reversal in the mother and daughter might have resulted from mutation or deletion of the SRY gene. When DNA from the mother (no DNA sample from the daughter was available) was tested by PCR with SRY-specific primers, no product was obtained. Likewise, PCR analysis for the Xp and Yp pseudoautosomal boundary revealed only the X-specific product. These results indicated that the Y chromosome resulted from an abnormal X-Y interchange with the breakpoint on Yp proximal to SRY, as found in about 10– 15 % of females with XY gonadal dysgenesis (Wolf et al., 1992). It has been shown that Xp-Yp abnormal interchange frequently occurs within the genomic regions of the closely related genes PRKX on Xp22.3 and PRKY on Yp11.2 (Schiebel et al., 1997b). We therefore analyzed the maternal DNA for the pres- Fig. 1. Fluorescence in situ hybridization of probes for ARSE (green) and SHOX (red) to metaphase spreads of lymphoblastoid cells of R.M. The position of the centromere is indicated by the white line on one X chromosome and on the Y chromosome. Note the Xp-specific ARSE signals close to the pseudoautosomal SHOX signals on all three sex chromosomes. The green signals on Yq derive from the ARSE pseudogene (see Fig. 2). ence of Y-specific sequences from the PRKY gene and its flanking regions by PCR. No PCR products were obtained for 3)flanking sequences (sY68); for exons 3, 2, and 1; and for the 5)-flanking primer pair TranY1; PCR was positive for the 5)flanking sequence TranD and for AMELY. These results place the breakpoint between TranY1 and TranD, between 16 kb and 44.5 kb upstream of the first PRKY exon (according to the sequence of BACs RP11-301O17 and RP11-558K21 (GenBank accession number NT_003007)), and thus within or near the translocation hot-spot region A (Schiebel et al., 1997b). To obtain further evidence for the presence of Xp material on the Y chromosome of R.M., FISH was performed. Twocolor FISH with probes for PRK and the pseudoautosomal SHOX gene revealed strong signals at the tips of the short arms of both X chromosomes and of the Y chromosome (not shown). As PRKY normally maps far from the Yp telomere in Yp11.2, near the centromere on the human Y chromosome, and as PRKX maps in Xp22.33 at a location that cannot be distinguished from the pseudoautosomal SHOX locus by FISH (Gläser et al., 1997a), these FISH mapping data are indicative of the presence of Xp material on the Y chromosome of R.M. Proof for this was obtained by two-color FISH with probes for SHOX and for the Xp-specific ARSE gene. As shown in Fig. 1, the ARSE probe gives hybridization signals (green) not only at the tip of both X chromosomes but also at the tip of the Y chromosome. These signals are always close to the signals (red) for the pseudoautosomal probe SHOX. In addition, hybridization signals are also seen on Yq, indicating the constitutive Y-chro- Cytogenet Cell Genet 91:204–207 (2000) 205 Fig. 2. Schematic of distal Xp22.3, of a normal Y chromosome, and of the Y chromosome of the XXY female R.M. The pseudoautosomal region is shaded light gray, the X-specific region dark gray; the Y-specific region is shown in white. The positions of the genes analyzed and of the X and Y pseudoautosomal boundaries PABX and PABY are indicated. The figure is only approximately to scale. „ARSE = ARSE pseudogene on Yq. mosomal position of the ARSE pseudogene (Meroni et al., 1996; Gläser et al., 1997b). These results indicate that during the aberrant X-Y interchange, the breakpoint on Xp must have occurred proximal to ARSE and PRKX that are located 150– 200 kb (Meroni et al., 1996) and approximately 1 Mb (Klink et al., 1995) from the Xp pseudoautosomal boundary, respectively. Taken together, our data show that the Y chromosome in R.M. and, by inference, in her daughter B.M., has the structure depicted schematically in Fig. 2: replacement of Yp material that includes SRY and PRKY by Xp material up to and including PRKX. The absence of SRY explains the sex reversal in these two XXY females. Discussion The Y chromosome of R.M. has the same general structure (Fig. 2) found in about 10–15 % of females with XY gonadal dysgenesis (Swyer syndrome). As in these cases, this structurally altered Y chromosome most likely resulted from an aberrant X-Y interchange during male meiosis, whereby Xp material was transferred to Yp with concomitant loss of Yp material. The breakpoint on Yp could be located to the vicinity of the PRKY gene, within or proximal to the recombination hot-spot region A, implying that the breakpoint on Xp occurred within the homologous region of the PRKX gene (Schiebel et al., 1997b). This unequal crossover between PRKY and PRKX must have occurred during meiosis in R.M.’s father or even in her maternal grandfather. A cytogenetic analysis of R.M.’s mother could answer this question. In 46,XY females with gonadal dysgenesis, the ovaries degenerate, losing oocytes at an early stage, and are present in adults as nonfunctional streaks of connective tissue (streak gonads). Such XY females are thus invariably sterile. In contrast, the XXY female R.M. is a fully fertile adult, with three 206 Cytogenet Cell Genet 91:204–207 (2000) children, one son and two daughters. It is well known that for maintenance of ovarian function during later fetal and postnatal development, two X chromosomes are needed, as also shown by the ovarian degeneration in 45,X females with Turner syndrome (Simpson and Rajkovic, 1999). The presence of the second X chromosome in R.M., likely due to maternal (i.e., grandmaternal) nondisjunction, prevents the infertility shown by XY gonadal dysgenesis females. R.M. is thus arguably the first human female described who is sex reversed in the fullest sense, being a completely normal female with respect to phenotypic, genital, and gonadal sex. Other than in the human, 39,X and Tdy (Sry)-negative XY female mice are both fertile, although fertility is reduced. In a comparative study of Tdy (Sry)-negative XY with Tdy (Sry)negative XXY female mice, the XY females were of poor fertility, although 5 of 12 females produced at least one offspring, whereas the XXY females had larger, more frequent litters and a longer reproductive lifespan. The findings in these sexreversed female mice thus also indicate a significant contribution to fertility by the second X chromosome, which could be attributed to a reduction of unpaired sex chromosomes as a cause of oocyte loss at pachytene (Mahadevaiah et al., 1993). Of the three children of R.M., we know the chromosome constitution only in her youngest daughter B.M., which is the same as her mother’s. From her normal female phenotype, it is apparent that B.M. inherited one X chromosome and the aberrant Y chromosome from her mother, and the second X chromosome from her father. Her elder sister, now at 5 yr of age, can theoretically have one of four karyotypes, due to nondisjunction of the sex chromosomes at maternal meiosis: 46,XX (i.e., normal female); 47,XXX (i.e., close-to-normal female); 47,XXY*, with Y* denoting the aberrant Y (i.e., the same karyotype as her mother and sister); or 46,XY* (i.e., a Swyer karyotype). The three possible karyotypes for the brother, now at 7 yr of age, are 46,XY, 47,XXY, or 47,XYY*, all with a normal or near-normal male phenotype during boyhood. It would be interesting of course, and important, to analyze these two sibs in the future. It has been calculated from the frequency of maternal nondisjunction of about 1 in 1,000 meioses and from the incidence of XX males, most of whom are Yp positive, of approximately 1 in 20,000 newborn males, that the coincidence of these events, resulting in a Yp-positive XXX male, might be expected once in 20 million male births (Annerén et al., 1987). One would assume that Yp-deleted XY females should occur at frequencies equal to Yp-positive XX males, as they are the mirror-image result of aberrant Xp-Yp interchange. However, they are relatively infrequent compared with the number of Yp-positive XX males, possibly due to embryonic lethal effects that may be associated with Yp deletions (Levilliers et al., 1989). Therefore, the incidence for a fertile Yp-deleted XXY female as described in this report can be expected to be significantly less than 1 in 20 million female births. Acknowledgements G.S. and K.S. would like to thank C. Zeschnigk and K. Meyer, respectively, for expert technical assistance. References Annerén G, Andersson M, Page DC, Brown LG, Berg M, Läckgren G, Gustavson K-H, de la Chapelle A: An XXX male resulting from paternal X-Y interchange and maternal X-X nondisjunction. Am J hum Genet 41:594–604 (1987). Bartsch-Sandhoff M, Stephan L, Röhrborn G, Pawlowitzki IH, Scholz W: Ein Fall von testiculärer Feminisierung mit dem Karyotyp 47,XXY. Hum Genet 31:59–65 (1976). Franco B, Meroni G, Parenti G, Levilliers J, Bernard L, Gebbia M, Cox L, Maroteaux P, Sheffield L, Rappold GA, Andria G, Petit C, Ballabio A: A cluster of sulfatase genes on Xp22.3: mutations in chondrodysplasia punctata (CDPX) and implications for warfarin embryopathy. Cell 81:15–25 (1995). Gerli M, Migliorini G, Bocchini V, Venti G, Ferrarese R, Donti E, Rosi G: A case of complete testicular feminization and 47,XXY karyotype. J med Genet 16:480–483 (1979). German J, Vesell M: Testicular feminization in monozygotic twins with 47 chromosomes (XXY). Annls Génét 9:5–8 (1966). Gläser B, Grützner F, Taylor K, Schiebel K, Meroni G, Tsioupra K, Pasantes J, Rietschel W, Toder R, Willmann U, Zeitler S, Yen P, Ballabio A, Rappold G, Schempp W: Comparative mapping of Xp22 genes in hominoids: evolutionary linear instability of their Y homologues. Chrom Res 5:167–176 (1997a). Gläser B, Hierl T, Taylor K, Schiebel K, Zeitler S, Papadopoullos K, Rappold G, Schempp W: Highresolution fluorescence in situ hybridization of human Y-linked genes on released chromatin. Chrom Res 5:23–30 (1997b). Hook EB, Hamerton JL: The frequency of chromosome abnormalities detected in consecutive newborn studies, in Hook EB, Porter IH (eds): Population Cytogenetics, pp 63–79 (Academic Press, New York 1977). Jacobs PA, Hassold TJ, Whittington E, Butler G, Collyer S, Keston M, Lee M: Klinefelter’s syndrome: an analysis of the origin of the additional sex chromosome using molecular probes. Ann hum Genet 52:93–109 (1988). Jacobs PA, Strong JA: A case of human intersexuality having a possible XXY sex determining mechanism. Nature 183:302–303 (1959). Jäger RJ, Anvret M, Hall K, Scherer G: A human XY female with a frame shift mutation in the candidate testis-determining gene SRY. Nature 348: 452–454 (1990a). Jäger RJ, Ebensperger C, Fraccaro M, Scherer G: A ZFY-negative 46,XX true hermaphrodite is positive for the Y pseudoautosomal boundary. Hum Genet 85:666–668 (1990b). Klink A, Schiebel K, Winkelmann M, Rao E, Horsthemke B, Lüdecke HJ, Claussen U, Scherer G, Rappold G: The human protein kinase gene PKX1 on Xp22.3 displays Xp/Yp homology and is a site of chromosomal instability. Hum molec Genet 4:869–878 (1995). Levilliers J, Quack B, Weissenbach J, Petit C: Exchange of terminal portions of X- and Y-chromosomal short arms in human XY females. Proc natl Acad Sci, USA 86:2296–2300 (1989). Mahadevaiah SK, Lovell-Badge R, Burgoyne PS: Tdynegative XY, XXY and XYY female mice: breeding data and synaptonemal complex analysis. J Reprod Fertil 97:151–160 (1993). Meroni G, Franco B, Archidiacono N, Messali S, Andolfi G, Rocchi M, Ballabio A: Characterization of a cluster of sulfatase genes on Xp22.3 suggests gene duplications in an ancestral pseudoautosomal region. Hum molec Genet 5:423–431 (1996). Müller U, Latt SA, Donlon T: Y-specific DNA sequences in male patients with 46,XX and 47,XXX karyotypes. Am J med Genet 28:393–401 (1987). Müller U, Schneider NR, Marks JF, Kupke KG, Wilson GN: Maternal meiosis II nondisjunction in a case of 47,XXY testicular feminization. Hum Genet 84:289–292 (1990). Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, Nordsiek G, Heinrich U, Breuning MH, Ranke MB, Rosenthal A, Ogata T, Rappold GA: Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nature Genet 16: 54–63 (1997). Ried T, Lengauer C, Cremer T, Wiegant J, Raap AK, van der Ploeg M, Groitl P, Lipp M: Specific metaphase and interphase detection of the breakpoint region in 8q24 of Burkitt lymphoma cells by triplecolor fluorescence in situ hybridization. Genes Chrom Cancer 4:69–74 (1992). Schempp W, Binkele A, Arnemann J, Gläser B, Ma K, Taylor K, Toder R, Wolfe J, Zeitler S, Chandley AC: Comparative mapping of YRRM- and TSPYrelated cosmids in man and hominoid apes. Chrom Res 3:227–234 (1995). Scherer G, Schempp W, Fraccaro M, Bausch E, Bigozzi V, Maraschio P, Montali E, Simoni G, Wolf U: Analysis of two 47,XXX males reveals X-Y interchange and maternal or paternal nondisjunction. Hum Genet 81:247–251 (1989). Schiebel K, Mertz A, Winkelmann M, Gläser B, Schempp W, Rappold G: FISH localization of the human Y-homolog of protein kinase PRKX (PRKY) to Yp11.2 and two pseudogenes to 15q26 and Xq12 → q13. Cytogenet Cell Genet 76:49–52 (1997a). Schiebel K, Winkelmann M, Mertz A, Xu X, Page DC, Weil D, Petit C, Rappold GA: Abnormal XY interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+)XX males and (Y–)XY females. Hum molec Genet 6:1985–1989 (1997b). Schmid M, Guttenbach M, Enders H, Terruhn V: A 47,XXY female with unusual genitalia. Hum Genet 90:346–349 (1992). Simpson JL, Rajkovic A: Ovarian differentiation and gonadal failure. Am J med Genet 89:186–200 (1999). Thangaraj K, Gupta NJ, Chakravarty B, Singh L: A 47,XXY female. Lancet 352:1121 (1998). Uehara S, Tamura M, Nata M, Kanetake J, Hashiyada M, Terada Y, Yaegashi N, Funato T, Yajima A: Complete androgen insensitivity in a 47,XXY patient with uniparental disomy for the X chromosome. Am J med Genet 86:107–111 (1999). Wolf U, Schempp W, Scherer G: Molecular biology of the human Y chromosome. Rev physiol biochem Pharmacol 121:147–213 (1992). Cytogenet Cell Genet 91:204–207 (2000) 207