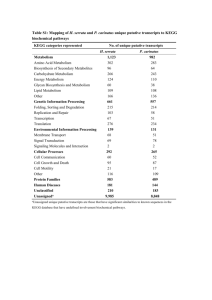
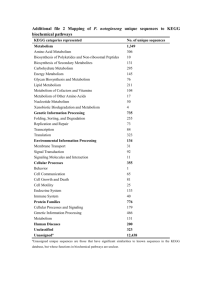
supplementary material Appendix I
advertisement

SPSS Step 1: Normality tests go to Analyze – Descriptive Statistics <select> Explore from the drop down list. In the box that opens enter the column identifier for the data that you wish to test in the Dependent List box. Click on Plots and tick the Normality plots with tests option. Click Continue then click Ok. The relevant output for this test can be found in the following table: Tests of Normality Kolmogorov-Smirnova Statistic Metabolism .140 Df Shapiro-Wilk Sig. 15 .200 Statistic * .957 df Sig. 15 .633 a. Lilliefors Significance Correction *. This is a lower bound of the true significance. The significance of the test is indicated by the p value in the table. If the p value is less than 0.05 then the distribution of data differs significantly from normal. If it is > .05 then the data can be considered normally distributed. A normality plot will also be shown called Normal Q-Q Plot of ‘column identifier’. EXAMINE VARIABLES=Metabolism /PLOT BOXPLOT STEMLEAF NPPLOT /COMPARE GROUPS /STATISTICS DESCRIPTIVES /CINTERVAL 95 /MISSING LISTWISE /NOTOTAL. Step 3: Attempt to normalise the distribution by transforming it. Data can be easily transformed by using the Transform – Compute Variable command. Enter a name for your new variable in the Target Variable box and enter your transformation in the Numeric Expression box (e.g., LG10(Variable name)). SPSS will create a new column with the transformed variable. COMPUTE LOGMetabolism=LG10(Metabolism). EXECUTE. Alternatively data may be transformed using the Box-Cox procedure Go to Transform – Prepare Data for Modelling <select> Automatic from the drop down list. In the Fields tab you can specify which variables to transform by moving them to the Inputs box. In the Settings tab click on Rescale Fields. Tick the box before ‘Rescale a continuous target with a Box-Cox transformation to reduce skew’. Click Run. This will create a new column with the transformed variable. Step 7: Paired comparisons of parametric normally distributed data For two sample t-test Go to Analyze – Compare Means <select> Independent Samples T-Test from the drop down list. This will open a new window. Data should be organized as shown in Table 1. Add the variable to be tested in the Test Variable(s) box. You can enter multiple variables (e.g., metabolism and body mass) if you want. Identify which data belong to which treatment by entering your treatment column into the Grouping Variable box. You now need to define your treatment groups by clicking the Define Groups button. A new window will pop up. When using numeric treatment groups enter the values for Group 1 and Group 2 (i.e., 1 and 2 respectively in our example in Table 1). If you have used string codes you need to identify the codes between apostrophe’s, e.g., for control = C and treatment =T, enter “C” for Group 1 and “T” for Group 2. Click Continue. Click Ok. The output appears as follows. Group Statistics Treatment Metabolism N Mean Std. Deviation Std. Error Mean 1.00 10 1.5380 .19510 .06169 2.00 5 .9760 .20403 .09125 Independent Samples Test Levene's Test for Equality of Variances t-test for Equality of Means 95% Confidence Interval Sig. Metabolism Equal df (2- Mean Std. Error tailed) Difference Difference of the Difference F Sig. t Lower Upper .348 .565 5.185 13 .000 .56200 .10839 .32784 .79616 5.102 7.771 .001 .56200 .11015 .30670 .81730 variances assumed Equal variances not assumed The first table summarizes your data and gives sample sizes, Mean, SD and SEM. The second table gives the results for the t-test. A t-test assumes equal variances and the first two columns in the table show the results for Levene’s test for equality of variances. If p>0.05 variances are equal and you will use the top row. The next 3 columns give the results for the independent t-test (tvalue, degrees of freedom and p-value respectively). In this case p<0.05 and the differences between the groups is statistically significant. T-TEST GROUPS=treatment(1 2) /MISSING=ANALYSIS /VARIABLES=Metabolism /CRITERIA=CI(.95). For Paired t-test Go to Analyze – Compare Means <select> Paired Samples T-Test from the drop down list. This will open a new window. Data should be organized as shown in Table 2. Add the column identifiers of interest into the Paired Variables box next to pair 1 (i.e., under variable1 and variable2). Click Ok.The output appears as follows. Paired Samples Correlations N Pair 1 Metabolism & Metabolism2 Correlation 15 .909 Sig. .000 Paired Samples Test Paired Differences t df Sig. (2- 95% Confidence Interval Mean Pair Metabolism - 1 Metabolism2 Std. Std. Error Deviation Mean -.12000 .14933 tailed) of the Difference Lower .03856 Upper -.20270 -.03730 -3.112 14 .008 Paired Samples Statistics Mean Pair 1 N Std. Deviation Std. Error Mean Metabolism 1.3507 15 .33401 .08624 Metabolism2 1.4707 15 .25018 .06460 The first table shows if there is a significant correlation between the two measurements of interest. In this case there is a significant correlation between both measurements of metabolism (p<0.01). This need not always be the case, and the results for the paired t-test are valid also if there is not a significant relationship between the variables of interest. The last three columns of the second table show the results for your paired t-test (t-value, degrees of freedom and p-value respectively). In this case p<0.05 and a significant effect of treatment is shown. The third table shows your descriptive statistics (mean, sd and sem). T-TEST PAIRS=Metabolism WITH Metabolism2 (PAIRED) /CRITERIA=CI(.9500) /MISSING=ANALYSIS. Step 8: multiple treatment levels, parametric tests For One Way ANOVA Go to Analyze – Compare Means <select> One way ANOVA from the drop down list. This will open a new window. Data should be organized as shown in Table 1. Add variable of interest (e.g., metabolism) to Dependent List box. Add column identifier for treatment levels to the Factor box (e.g., levels). Click Ok. The output appears as follows. ANOVA Metabolism Sum of Squares Between Groups Within Groups Total df Mean Square 1.342 2 .671 .220 12 .018 1.562 14 F 36.582 Sig. .000 The F and P values for the treatment effect and for the individual effect are shown in the last two columns of the table. A p-value < than .05 indicates a significant effect. In this example there was a significant treatment effect. ONEWAY Metabolism BY levels /MISSING ANALYSIS. For Repeated Measured ANOVA Data should be organized as shown in Table 2. Go to Analyze – General Linear Model <select> Repeated Measures from the drop down list. This will open a new window. Add the ‘Number of levels’ that you have in the appropriate box (i.e., the number of repeated measurements; i.e., in this case 3). Then click Add. Click Define. This will open a new window. Add the column identifiers of interest into the Within-Subjects Variables box. The number of levels that you have identified in the previous step will show up in the box and you need to add the column identifiers for each level. If you used different levels of treatments or other factor that are different between subjects like for instance sex, this can be added to the Between-Subjects Factor(s) box. Click Ok. The relevant output for this test can be found in the following tables: Tests of Within-Subjects Effects Measure:MEASURE_1 Type III Sum of Source Squares Treatment Error(Treatment) df Mean Square F Sig. Sphericity Assumed .109 2 .054 3.779 .035 Greenhouse-Geisser .109 1.799 .060 3.779 .041 Huynh-Feldt .109 2.000 .054 3.779 .035 Lower-bound .109 1.000 .109 3.779 .072 Sphericity Assumed .403 28 .014 Greenhouse-Geisser .403 25.189 .016 Huynh-Feldt .403 28.000 .014 Lower-bound .403 14.000 .029 Tests of Between-Subjects Effects Measure:MEASURE_1 Transformed Variable:Average Type III Sum of Source Intercept Error Squares Df Mean Square 89.183 1 89.183 2.802 14 .200 F 445.546 Sig. .000 The Tests of Within-Subject Effects Table shows the results for your repeated measures (e.g., metabolism measured at different time points in the same individual). In our example (Table 2) this refers to metabolism measured at control, treatment 1 and treatment 2. The first row (Spericity Assumed) shows if there was a significant effect of treatment (F1,28=3.8, p=0.035 in our example). If you added a Between-Subjects Factor (e.g., sex) the F and p-values will be shown in the last two columns of the Tests of Between-Subjects Effects Table. In this case, no value is shown because no factor was added. Note that sphericity is assumed in a RM ANOVA. A violation of sphericity occurs when the variances of the differences between all combinations of the groups are not equal and this is tested in the Mauchly's Test of Sphericity test for which the results are given as part of the SPSS output in a RM ANOVA (in Mauchly's Test of Sphericity table). When the probability of Mauchly's test statistic is less than or equal to 0.05 (i.e., p < .05), sphericity cannot be assumed and a correction needs to be made to the F and p-value. SPSS provides three corrections, Greenhouse-Geisser, Huynh-Feldt and Lower-bound. For more details about these tests we would refer you to a statistical text book. GLM Metabolism1 Metabolism2 Metabolism3 /WSFACTOR=Treatment 3 Polynomial /METHOD=SSTYPE(3) /CRITERIA=ALPHA(.05) /WSDESIGN=Treatment. Step 10: post hoc tests For One Way ANOVA Conduct the one-way ANOVA as described under section 9. However before clicking on OK, click on the button labelled Post Hoc. This will open a new window. Choose the test you want to use by ticking the appropriate box, e.g., Tukey test. Click Continue and click OK. This time in addition to the ANOVA results there is an additional output below the analysis of variance table as follows. Post Hoc Tests Multiple Comparisons Metabolism Tukey HSD 95% Confidence Interval Mean Difference (I) levels 1.00 2.00 (J) levels Std. Error Sig. Lower Bound Upper Bound 2.00 .34000 * .08565 .005 .1115 .5685 3.00 .73200* .08565 .000 .5035 .9605 1.00 -.34000 * .08565 .005 -.5685 -.1115 .39200 * .08565 .002 .1635 .6205 1.00 -.73200 * .08565 .000 -.9605 -.5035 2.00 -.39200* .08565 .002 -.6205 -.1635 3.00 3.00 (I-J) Multiple Comparisons Metabolism Tukey HSD 95% Confidence Interval Mean Difference (I) levels (J) levels 1.00 2.00 .34000* .08565 .005 .1115 .5685 3.00 .73200 * .08565 .000 .5035 .9605 -.34000 * .08565 .005 -.5685 -.1115 3.00 .39200* .08565 .002 .1635 .6205 1.00 -.73200 * .08565 .000 -.9605 -.5035 -.39200 * .08565 .002 -.6205 -.1635 2.00 (I-J) 1.00 3.00 2.00 Std. Error Sig. Lower Bound Upper Bound *. The mean difference is significant at the 0.05 level. Homogeneous Subsets Metabolism Tukey HSD a Subset for alpha = 0.05 Levels N 1 3.00 5 2.00 5 1.00 5 Sig. 2 3 .9760 1.3680 1.7080 1.000 1.000 1.000 Means for groups in homogeneous subsets are displayed. a. Uses Harmonic Mean Sample Size = 5.000. The Multiple Comparisons table shows pair-wise comparisons for the different levels of treatment. P values<0.05 between groups indicate the groups differ significantly. The Homogeneous Subsets table summarises the results from the multiple comparisons and shows the mean values for the different levels of treatment and whether they differ or not. In this case metabolism is different between all levels of treatment. ONEWAY Metabolism BY levels /MISSING ANALYSIS /POSTHOC=TUKEY ALPHA(0.05) For Repeated Measures ANOVA Conduct the Repeated Measures ANOVA as described under section 9. However before clicking on OK, click on the button labelled Post Hoc. This will open a new window. Add the Factor of interest to the ‘Post Hoc Tests For’ box. Choose the test you want to use by ticking the appropriate box, e.g., Tukey test. Click Continue and click OK. This time in addition to the ANOVA results there is an additional output below the analysis of variance that is similar to the output shown above for the OneWay ANOVA. GLM Metabolism Metabolism2 BY levels /WSFACTOR=Time 2 Polynomial /METHOD=SSTYPE(3) /POSTHOC=levels(TUKEY) /CRITERIA=ALPHA(.05) /WSDESIGN=Time /DESIGN=levels. Step 11 SPSS does not provide the capability to perform power analysis. Alternative programs need to be used instead. Step 13 : two way anova For Two-way ANOVA Data should be organized as shown in Table 1. Go to Analyze – General Linear Model <select> Univariate from the drop down list. This will open a new window. Add your variable to be tested to the Dependent Variable box (e.g., metabolism). Add your fixed factors to the Fixed Factor(s) box (e.g., treatment and sex). The output appears as follows. Between-Subjects Factors N Sex Levels .00 9 1.00 6 1.00 5 2.00 5 3.00 5 Tests of Between-Subjects Effects Dependent Variable:Metabolism1 Type III Sum of Source Squares df Mean Square F Sig. Corrected Model 1.417 a 5 .283 17.596 .000 Intercept 26.645 1 26.645 1654.406 .000 .033 1 .033 2.065 .185 1.210 2 .605 37.577 .000 Sex * Levels .042 2 .021 1.300 .319 Error .145 9 .016 Total 28.926 15 1.562 14 Sex Levels Corrected Total a. R Squared = .907 (Adjusted R Squared = .856) The last two columns of the Tests of Between-Subjects Effects table show the F and p-values. In this case there was a significant effect of treatment level (p<0.001), but there was no significant effect of sex (p=0.185) and no significant sex by treatment interaction (p=0.319). A significant interaction effect implies that both sexes responded differently to the treatment (e.g., one sex in or decreased more than the other). For Repeated Measures Two-way ANOVA Data should be organised as shown in Table 2. As Repeated measures one-way ANOVA (see 9), but add a Between-Subjects Factor (e.g., sex). In the output the extra factor will be included in both the Tests of Within and between effects tables. Step 14: Same as 13 for Two-way ANOVA, but add extra fixed factor(s) in the Fixed Factor(s) box. Factors can be added to or removed from the model by using the Model button. This will open a new window. Tick Custom and add/remove variables of interest. Step 18: non parametric tests paired comparisons Mann whitney U-test Go to Analyze – Nonparametric tests – Legacy dialogs <select> Two independent samples from the drop down list. This will open a new window. Data should be organized as shown in Table 1. Add variable to be tested to the Test Variable list box (e.g., metabolism). Identify which data belong to which treatment by entering your treatment column into the Grouping Variable box. You now need to define your treatment groups by clicking the Define Groups button. A new window will pop up. When using numeric treatment groups enter the values for Group 1 and Group 2 (i.e., 1 and 2 respectively in our example in Table 1). If you have used string codes you need to identify the codes between apostrophe’s, e.g., for control = C and treatment =T, enter “C” for Group 1 and “T” for Group 2. Click Continue. Make sure the box before Mann-Whitney U is ticked. Click Ok. The output appears as follows. Ranks treatment Metabolism N Mean Rank Sum of Ranks 1.00 10 10.50 105.00 2.00 5 3.00 15.00 Total 15 Test Statisticsb Metabolism Mann-Whitney U .000 Wilcoxon W 15.000 Z -3.067 Asymp. Sig. (2-tailed) .002 Exact Sig. [2*(1-tailed Sig.)] .001a a. Not corrected for ties. b. Grouping Variable: treatment Z and p-value (Asump. Sig. (2-tailed)) are shown in the Test statistics table. NPAR TESTS /M-W= Metabolism BY treatment(1 2) /MISSING ANALYSIS. Wilcoxon matched pairs test Go to Analyze – Nonparametric Tests – Legacy dialogs <select> Two Related Samples from the drop down list. This will open a new window. Data should be organized as shown in Table 2. Add the column identifiers of interest into the Test Pairs box next to pair 1 (i.e., under variable1 and variable2). Make sure the box before Wilcoxon in ticked. Click Ok. The output appears as follows. Ranks N Metabolism2 - Metabolism Mean Rank Sum of Ranks Negative Ranks 3 a 4.00 12.00 Positive Ranks 12b 9.00 108.00 c Ties 0 Total 15 a. Metabolism2 < Metabolism b. Metabolism2 > Metabolism c. Metabolism2 = Metabolism Test Statisticsb Metabolism2 – Metabolism -2.728a Z Asymp. Sig. (2-tailed) .006 a. Based on negative ranks. b. Wilcoxon Signed Ranks Test Z and p-value (Asump. Sig. (2-tailed)) are shown in the Test statistics table. NPAR TESTS /WILCOXON=Metabolism WITH Metabolism2 (PAIRED) /MISSING ANALYSIS. Step 19 non parametric analysis when there are multiple treatments or levels: Kruskal-Wallis ANOVA Go to Analyze – Nonparametric tests – Legacy dialogs <select> k independent samples from the drop down list. This will open a new window. Data should be organized as shown in Table 1. Add variable to be tested to the Test Variable list box (e.g., metabolism). Identify which data belong to which treatment by entering your treatment column into the Grouping Variable box. You now need to define your treatment groups by clicking the Define Range button. A new window will pop up. Enter Minimum and Maximum values for your groups (i.e., 1 - 3 respectively for the treatment levels in our example in Table 1). Click Continue. Make sure the box before Kruskal-Wallis H is ticked. Click Ok. The output appears as follows. Ranks levels Metabolism N Mean Rank 1.00 5 13.00 2.00 5 8.00 3.00 5 3.00 Total 15 Test Statisticsa,b Metabolism Chi-Square 12.545 Df 2 Asymp. Sig. .002 a. Kruskal Wallis Test b. Grouping Variable: levels X2 and p-value (Asump. Sig. (2-tailed)) are shown in the Test Statistics table. NPAR TESTS /K-W=Metabolism BY levels(1 3) /MISSING ANALYSIS. Repeated measures Friedman Test Go to Analyze – Nonparametric Tests – Legacy dialogs <select> k Related Samples from the drop down list. This will open a new window. Data should be organized as shown in Table 2. Add the column identifiers of interest into the Test Variables. Click Ok. The output appears as follows. Ranks Mean Rank Metabolism 1.20 Metabolism2 1.80 Test Statisticsa N Chi-Square Df Asymp. Sig. 15 5.400 1 .020 a. Friedman Test X2 and p-value (Asump. Sig. (2-tailed)) are shown in the Test Statistics table. Step 23 Analysis of Covariance: ANCOVA Go to Analyze – General Linear Model <select> Univariate from the drop down list. This will open a new window. Data should be organized as shown in Table 1. Add your variable to be tested to the Dependent Variable box (e.g., metabolism). Add the column identifier for treatments to the Fixed Factor(s) box (e.g., levels) and add the covariate (e.g., body mass) to the Covariate box. The output appears as follows. Tests of Between-Subjects Effects Dependent Variable: Metabolism Source Type III Sum of df Mean Square F Sig. Squares a 3 .468 32.441 .000 Intercept .001 1 .001 .065 .804 Bodymass .061 1 .061 4.264 .063 1.325 2 .663 45.957 .000 Error .159 11 .014 Total 28.926 15 1.562 14 Corrected Model Levels Corrected Total 1.403 a. R Squared = .898 (Adjusted R Squared = .871) F and P-values are shown in the final two columns. In this example the bodyweight effect did not reach statistical significance (p=0.063), but there was a significant effect of treatment (p<0.05). In SPSS the default full factorial model does not include an interaction effect between body mass and treatment. To include this interaction rerun the analysis (go to Analyze – General Linear Model <select> Univariate from the drop down list) and click the Model button. This will open a new window. Tick the Custom box and then click and add factors as appropriate using the arrow button. To add an interaction effect, select two factors (in this case body mass and levels) and click the arrow button and the interaction effect will show up in the right box (i.e., levels x body mass). The output is as follows. Tests of Between-Subjects Effects Dependent Variable: Metabolism Source Type III Sum of df Mean Square F Sig. Squares a 5 .288 21.138 .000 Intercept .004 1 .004 .288 .604 Levels .056 2 .028 2.055 .184 Bodymass .077 1 .077 5.644 .042 Levels * Bodymass .036 2 .018 1.323 .314 Error .123 9 .014 Total 28.926 15 1.562 14 Corrected Model 1.439 Corrected Total a. R Squared = .922 (Adjusted R Squared = .878) Note that when the interaction effect is included there is no significant effect of treatment level. The interaction effect is also not significant. In this case you would remove the interaction effect and analyse the data without the interaction effect as shown above. Step 37 Correlation matrix Select Analyse <select> Correlation and <select> Bivariate. This will open a new window. Add variables of interest to the variables box. Tick relevant box under correlation coefficients (i.e., Pearson). The output is a correlation matrix. A typical matrix might be as follows for a situation where 5 organ weights are available. BAT 0.13 Skeletal muscle 0.66 0.17 0.55 0.93 1.00 0.11 0.03 Organ Liver WAT Brain Liver 1.00 0.32 1.00 WAT Brain 0.22 Skeletal muscle 1.00 0.61 This table highlights that WAT and BAT are highly correlated and hence not independent predictor variables. Skeletal muscle also is quite highly correlated to the liver mass. One can proceed with the analysis ignoring these effects but one should be aware that such correlations may compromise the outcome. In this case a strong effect of WAT might emerge because of the effect of BAT on metabolism combined with the high correlation of WAT with BAT. This analysis requires the number of observations (i.e., individuals) to exceed by at least a factor of 3 the number of predictor variables included into the analysis. Hence in this situation one would have 5 predictors so for each group (i.e., treatment levels and control) one would need at least 15 individuals – and preferably many more. Interpretation of these effects depends on the complexity of the interactions. The bottom line is to diagnose an overall treatment effect controlling for these body composition variables. If there is an overall treatment effect one can establish where this occurs using the multiple range tests (TUKEY TEST and DUNCAN’S MULTIPLE RANGE TEST). Step 38: PRINCIPAL COMPONENTS ANALYSIS. Principal Component Analysis can be performed using SPSS, but the procedure to do so is hidden within the procedure for factor analysis. Go to Analyze <select> Dimension Reduction <select> Factor. This opens a new window. Add the column identifiers of the variables of interest to the Variables box. Click the Extraction button. This will open a new window. Under Method select Principal Components using the drop-down menu. Tick Correlation matrix and Unrotated factor solution. To restrict the number of components tick Fixed number of Factors and type the number of components you want in the Factors to extract box (e.g., 5). Click Continue this will take you back to the first window. Click Ok. The output is as follows. Communalities Initial Extraction Carcass 1.000 .924 HEART 1.000 .874 LIVER 1.000 .784 KIDNEY 1.000 .926 BRAIN 1.000 .672 Brown Fat 1.000 .861 Abdominal Fat 1.000 .926 Gonadal Fat 1.000 .939 Mesenteric Fat 1.000 .836 Gonads 1.000 .914 Large Intestine (g) 1.000 .807 Small Intestine (g) 1.000 .735 Stomach 1.000 .805 Lungs 1.000 .911 Pancreas 1.000 .839 Pelage 1.000 .942 Tail 1.000 .895 Extraction Method: Principal Component Analysis. Total Variance Explained Initial Eigenvalues Component Total % of Variance Extraction Sums of Squared Loadings Cumulative % Total % of Variance Cumulative % 1 7.688 45.221 45.221 7.688 45.221 45.221 2 3.430 20.176 65.396 3.430 20.176 65.396 3 1.614 9.493 74.889 1.614 9.493 74.889 4 1.060 6.235 81.123 1.060 6.235 81.123 5 .798 4.696 85.819 .798 4.696 85.819 6 .648 3.815 89.634 7 .550 3.237 92.871 8 .354 2.083 94.953 9 .283 1.663 96.616 10 .180 1.061 97.677 11 .125 .735 98.412 12 .103 .605 99.018 13 .075 .440 99.458 14 .046 .272 99.731 15 .023 .134 99.865 16 .019 .110 99.974 17 .004 .026 100.000 Extraction Method: Principal Component Analysis. Component Matrixa Component 1 2 3 4 5 Carcass .923 -.198 -.090 -.145 .069 HEART .655 .530 .067 .212 .337 LIVER .785 -.399 .021 -.017 .089 KIDNEY .800 .501 .113 -.035 -.145 BRAIN .373 .687 .229 -.090 .012 -.639 -.218 .615 .021 -.161 Abdominal Fat .625 -.617 .178 .335 -.106 Gonadal Fat .743 -.472 .140 .287 -.250 Mesenteric Fat .796 -.179 .310 .202 .182 Gonads .386 -.559 -.186 -.465 .449 Large Intestine (g) .869 -.025 -.124 .009 .187 Brown Fat Small Intestine (g) .412 .107 .730 -.132 .059 Stomach .370 .589 .430 -.368 -.027 -.052 .689 -.145 .586 .264 Pancreas .765 .223 -.320 -.068 -.312 Pelage .909 -.283 -.054 .056 -.173 Tail .635 .518 -.340 -.168 -.284 Lungs Extraction Method: Principal Component Analysis. a. 5 components extracted. MINITAB Step 1: Normality tests Go to statistics tab, <select> basic statistics from the drop down list <select> normality test (second from bottom). In the box that opens enter the column identifier for the data that you wish to test. Click on the actual test you wish to perform (e.g. Anderson-Darling test). A typical output for this test looks like the following for the data in table 1 column 2 Probability Plot of Metabolism Normal 99 Mean StDev N AD P-Value 95 90 1.351 0.3340 15 0.239 0.732 Percent 80 70 60 50 40 30 20 10 5 1 0.50 0.75 1.00 1.25 1.50 Metabolism 1.75 2.00 2.25 The significance of the test is indicated by the p value in the box to the right of the plot. If the p value is less than 0.01 then the distribution of data differs significantly from normal. If it is > .01 then the data can be considered normally distributed. Step 3: Attempt to normalise the distribution by transforming it. Data can be easily transformed by going to Calc - Calculator. This will open a window. Enter a name for your new variable in the Store results in Variable box and enter your transformation in the Expression box (e.g., LOGTEN(Column Identifier)). Click Ok. Minitab will create a new column with the transformed variable. Alternatively data can be transformed using the box-cox procedure. Go to the statistics tab. Select Control charts from the dropdown box. Select BOXCOX from the options that appear. This opens a new window. Type the column identifier in the box you want to transform. Insert the number 1 in the box that says sub-group size. Click on options. In the new window that appears type a column identifier (e.g. C9 for column 9) in the box that says ‘store transformed data in’ where you want the transformed data to be stored. Click on OK. Closes window. Click on OK. Perfoms analysis. A typical output looks like this. Box-Cox Plot of BEE Lower CL Upper CL Lambda 1.3 (using 95.0% confidence) Estimate 0.88 Lower CL Upper CL 1.2 StDev Rounded Value -0.00 1.91 1.00 1.1 1.0 0.9 Limit 0.8 -5.0 -2.5 0.0 Lambda 2.5 5.0 The plot shows the optimal transformation value (lambda). The transformed data will now be in the column you specified in the options. Step 7: Paired comparisons of parametric normally distributed data For two sample t-test Go to the statistics tab and select ‘basic statistics’ from the drop down tab. Select 2t two sample t… from the available options and click on it. Opens new window. If you have formatted the data as detailed in the Table 1 then the data you are testing will be in one column (e.g., in the above example the energy expenditure data is in column 2) and the codes identifying which data are treatment and which control are in another column (in the above example column 4). In the new window select the ‘data in one column’ button and enter C2 in the data box and c4 in the subscripts box. Typical output (for analysis of metabolism against treatment group in Table one) looks as follows: Two-sample T for Metabolism Treatment 1 2 N 10 5 Mean 1.538 0.976 StDev 0.195 0.204 SE Mean 0.062 0.091 Difference = mu (1) - mu (2) Estimate for difference: 0.562 95% CI for difference: (0.302, 0.822) T-Test of difference = 0 (vs not =): T-Value = 5.10 DF = 7 P-Value = 0.001 The t-value and p value are shown on the bottom line. If P < .05 then the difference between the two groups is significant. Data for mean, sd and se for each of the treatment groups is shown in the table. In this case (metabolism data from table 1) there was a significant difference between treatment and control groups. For Paired t-test To use the paired t-test the data in Minitab needs to be organised as shown in Table 2, i.e., the data we are interested in testing for the treatment needs to be placed in a separate column from the control data and data from the same individual needs to be aligned in the same row. Go to the statistics tab. Select ‘basic statistics’ from the dropdown box. This opens a new window. Select t..t paired t test. Opens new window. Click the ‘samples in columns’ button. Enter the column identifiers into the two boxes. Click OK. Typical output (for data in table 2 comparing metabolism 1 and metabolism 2) looks as follows: Paired T for Metabolism1 - Metabolism2 Metabolism1 Metabolism2 Difference N 15 15 15 Mean 1.3507 1.4707 -0.1200 StDev 0.3340 0.2502 0.1493 SE Mean 0.0862 0.0646 0.0386 95% CI for mean difference: (-0.2027, -0.0373) T-Test of mean difference = 0 (vs not = 0): T-Value = -3.11 0.008 P-Value = The t-value and p value are shown on the bottom line. If P < .05 then there is a difference between the treatment and control. Sign of the t and the values in the table indicate the direction of the difference. In this case the difference is highly significant (P <.01) and the metabolism 1 is lower than metabolism 2. Step 8: multiple treatment levels, parametric tests For One way ANOVA Go to the statistics tab. <Select> ANOVA from the drop-down box. If the data are all in a single column with the identifiers for them in a second column then <Select> ‘one way...’ from the options. This opens a new window. In the response box type the identifier for the variable being tested (e.g., metabolism). In the factor box type the column that contains the treatment levels. On the other hand if the data are structured as in Table 2 with each measurement in a separate column <Select> ‘one way (unstacked)...’. This opens a new window. Enter the column identifiers for the columns containing the data into the box marked ‘Responses (in separate columns)’. Then click on OK. Using the data from Table 1 the output appears as follows: One-way ANOVA: Metabolism versus Levels Source Levels Error Total DF 2 12 14 SS 1.3418 0.2201 1.5619 S = 0.1354 Level 1 2 3 N 5 5 5 MS 0.6709 0.0183 F 36.58 R-Sq = 85.91% Mean 1.7080 1.3680 0.9760 StDev 0.0769 0.0864 0.2040 P 0.000 R-Sq(adj) = 83.56% Individual 95% CIs For Mean Based on Pooled StDev ------+---------+---------+---------+--(----*-----) (-----*----) (----*----) ------+---------+---------+---------+--1.00 1.25 1.50 1.75 Pooled StDev = 0.1354 F and P values are shown in the variance table at the start of the output. If the P value is less than .05 then there is a significant effect of the treatment. In this case there is a highly significant treatment effect. (Note Minitab refers to p values less than .001 as 0.000. These should be cited as P < .001). Repeated measures ANOVA: There is no specific procedure in MINITAB to perform a repeated measures ANOVA. The best way to perform this test is to use the general linear model test and include individual ID as a random factor in the model. The data needs to be in the ‘stacked format’ for this analysis – ie all the data need to be in a single column with other columns identifying the treatment and the individual IDs. The following analysis uses the data from Table 2 where 15 individuals are measured in 3 conditions (control and 2 treatments labelled metabolism1, 2 and 3) To perform this test, go to the statistics tab. <Select> ANOVA. From the options that appear <select> GLM – general linear model. This opens a new window. In the box that opens type the column identifier for the variable you are interested in testing into the response box. In the model box you need to enter the column identifier for the treatment levels and the column identifier for the column containing the individual IDs. In the box marked ‘random factors’ enter the same column identifier for the IDs. (Note if each individual is measured only once in each condition an interaction of individual and treatment level cannot be tested). The output appears as follows. General Linear Model: Metabolism versus ID, Treatment Factor Type ID random trmt fixed Levels Values 15 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 3 Metabolism1, Metabolism2, Metabolism3 Analysis of Variance for C21, using Adjusted SS for Tests Source DF Seq SS Adj SS Adj MS F P ID trmt Error Total 14 2 28 44 2.80231 0.10875 0.40292 3.31398 S = 0.119958 2.80231 0.10875 0.40292 R-Sq = 87.84% 0.20017 0.05438 0.01439 13.91 3.78 0.000 0.035 R-Sq(adj) = 80.89% The F and P values for the treatment effect and for the individual effect are shown in the variance table. A value less than .05 indicates a significant effect. In this example there was both a significant treatment effect, and also a significant individual effect. Step 10: post hoc tests For paired t-test see procedure detailed above in section 8. For post hoc tests proceed as follows. Conduct the one-way ANOVA as described under step 8. However before clicking on OK, click on the button labelled comparisons. Choose the test you want to use e.g. Tukey test. This time in addition to the ANOVA results there is an additional output below the analysis of variance table as follows. Grouping Information Using Tukey Method Levels 1 2 3 N 5 5 5 Mean 1.7080 1.3680 0.9760 Grouping A B C Means that do not share a letter are significantly different. Tukey 95% Simultaneous Confidence Intervals All Pairwise Comparisons among Levels of Levels Individual confidence level = 97.94% Levels = 1 subtracted from: Levels 2 3 Lower -0.5683 -0.9603 Center -0.3400 -0.7320 Upper -0.1117 -0.5037 ----+---------+---------+---------+----(----*-----) (-----*----) ----+---------+---------+---------+-----0.80 -0.40 -0.00 0.40 Levels = 2 subtracted from: Levels 3 Lower -0.6203 Center -0.3920 Upper -0.1637 ----+---------+---------+---------+----(-----*-----) ----+---------+---------+---------+-----0.80 -0.40 -0.00 0.40 The first part of this output shows the pairwise comparisons of each level. Inthis case the 3 groups all differ significantly from each other which is indicated in the table by the fact none of them share a letter adjacent to the level identifier. The information under the table shows the pairwise differences and their confidence limits. For repeated measures ANOVA tested in GLM Repeat the analysis as specified above under step 8 but before clicking on OK to run the test click on ‘comparisons’. In the new window that opens enter the column identifier for the treatment in the box labelled ‘terms’. Select the test required, e.g. Tukey test. The output appears as follows. Grouping Information Using Tukey Method and 95.0% Confidence C22 Metabolism2 Metabolism3 Metabolism1 N 15 15 15 Mean 1.5 1.4 1.4 Grouping A A B B Means that do not share a letter are significantly different. In this instance metabolism 1 doesn’t differ from metabolism 3 but it does differ from metabolism 2. However metabolism 2 and 3 are also not significantly different. Step 11 Power analysis Go to the statistics tab. Select ‘power analysis and sample size’. Select the test you used from the options that appear. Under each of the options you need to specify all the values except ‘power’, e.g., under two sample t-test you need to fill in the boxes that specify sample sizes, differences and standard deviation. The sample size is the number of measurements in each group. The difference is the size of the effect that you would consider important to detect. For example if you felt a difference would between groups would need to be 5% or larger before you would consider it important then you need to take the mean value across all the measurements and calculate 5% of that value. Finally add the pooled standard deviation from the output of the test. For example in the 2 sample t test detailed above the overall mean was 1.368 so 5% of this would be 0.0684. The standard deviation was 0.133 and the sample size per group was 10. Putting these into the respective boxes and clicking Ok runs the analysis. The typical output looks like this Power and Sample Size 2-Sample t Test Testing mean 1 = mean 2 (versus not =) Calculating power for mean 1 = mean 2 + difference Alpha = 0.05 Assumed standard deviation = 0.133 Difference 0.068 Sample Size 10 Power 0.191413 The sample size is for each group. The power for this example is 0.191 multiply this by 100 to express it as a %. So the power in this case to detect a 5% difference between means was only 19.1%. this means we cannot be sure that the absence of a difference wasn’t just a type 2 error because of the low sample size. If you run the test again but this time put the desired minimum power into the power box – i.e. 0.8, and leave the sample size box empty, this analysis would show that to detect a 5% difference would need 62 animals per group. Step 13 : two way anova Two-way ANOVA In Mintab the best way to perform Two way ANOVA is to use the general linear model (GLM) as detailed in section 9 above. Although there is a facility to do twoway ANOVA in MINITAB under the statistics and ANOVA tabs this only works for completely balanced designs. To perform two-way ANOVA by GLM the data needs to be in the ‘stacked format’ – i.e. all the response data need to be in a single column with other columns identifying the treatment or factor variables and individual IDs. To select this test choose <statistics>, <ANOVA>, <general linear model>. In the box labelled ‘responses’ type the column identifier for the variable you want to analyse (e.g. Metabolism). In the box labelled ‘model’ it is necessary to include the column identifiers for both of the treatment variables plus an additional term which is the multiplication of these two variables to reflect the interaction of the predictors. For example using the example of the data in Table 1. The treatment levels are in column 5 (C5) and the sex identifiers in column six (C6). Hence the model would be C5 C6 C5*C6 In the case of the metabolism data detailed in table 1 above with treatment and sex as factors the output appears as follows General Linear Model: Metabolism versus Treatment, Sex Factor Levels Sex Type fixed fixed Levels 3 2 Values 1, 2, 3 0, 1 Analysis of Variance for Metabolism, using Adjusted SS for Tests Source Levels Sex Levels*Sex Error Total DF 2 1 2 9 14 Seq SS 1.34181 0.03325 0.04188 0.14495 1.56189 S = 0.126908 R-Sq = 90.72% Adj SS 1.21041 0.03325 0.04188 0.14495 Adj MS 0.60520 0.03325 0.02094 0.01611 F 37.58 2.06 1.30 P 0.000 0.185 0.319 R-Sq(adj) = 85.56% The significance of the different effects is shown in the ANOVA table. In this case there is a significant treatment effect (p < 0.001), no significant sex effect (p > 0.05) and no significant sex by treatment interaction (p = 0.319). Repeated measures 2-way ANOVA To perform this test one simply repeats the above procedure but including a column with the individual IDs in it and put this into the model. This column is also entered into the box labelled ‘random factors’. Step 14: Proceed in the same way as for step 13 Two-way ANOVA. Add additional factors and interactions into the ‘model’ box. Step 18: non parametric tests paired comparisons Mann Whitney U-test The data need to be organised with the data for treatment and control in different columns. Using the data from table 1. Go to Statistics – nonparametrics – Mann Whitney. This opens a new window. Enter the column identifier for the treatment data and control data in the respective boxes. Click on OK. The output appears as follows. Mann-Whitney Test and CI: Treatment, Control T C N 10 5 Median 1.5650 1.0000 Point estimate for ETA1-ETA2 is 0.5600 95.7 Percent CI for ETA1-ETA2 is (0.2599,0.7799) W = 105.0 Test of ETA1 = ETA2 vs ETA1 not = ETA2 is significant at 0.0027 The test is significant at 0.0026 (adjusted for ties) The p value for the test is shown on the bottom line of the output. If this value is < .05 the difference between the columns is significant. Wilcoxon matched pairs The data need to be organised as in table 2. Before running the test it is necessary to subtract one column from the other. To do this go to Calc, select calculator. Identify the column where you wish the result to be placed (e.g. C10). Pt the subtraction calculation into the ‘expression’ box. Ie using the data in table 2 the expression would be ‘C3-C2’ (i.e. treatment minus control data) Go to Statistics – nonparametrics – 1-Sample Wilcoxon. This opens a new window. Put the column identifier for the column that contains the differences into the box marked ‘variables. Click the button marked ‘test median’ and enter the value 0.0 into the box if it doesn’t appear automatically. The output appears as follows. Wilcoxon Signed Rank Test: C10 Test of median = 0.000000 versus median not = 0.000000 C10 N 15 N for Test 15 Wilcoxon Statistic 108.0 P 0.007 Estimated Median 0.1075 The value for the Wilcoxon statistic and the associated p value are displayed in the table. If the P value is less than 0.05 as it is in this case there is a significant difference between the treatment and control. Step 19 non parametric analysis when there are multiple treatments or levels: Kruskal Wallis ANOVA Go to statistics- select nonparametrics- select Kruskal Wallis. In the response box enter the column identifier for the dependent variable eg metabolism and in the factor box type the identifier for the treatment variable. Click OK. The output appears as follows. Kruskal-Wallis Test: Metabolism versus Treatment Kruskal-Wallis Test on Metabolism Treatment 1 2 Overall H = 9.38 H = 9.41 N 10 5 15 Median 1.565 1.000 DF = 1 DF = 1 Ave Rank 10.5 3.0 8.0 P = 0.002 P = 0.002 Z 3.06 -3.06 (adjusted for ties) The significance is indicated on the last line as a P value. If P < .05 there is a significant treatment effect Friedman test To apply the Friedman test in Minitab the data needs to be structured in a different way. All the data needs to be in a single column with the individual identifiers in a separate column and the treatments in a third column. To generate these columns from the data in table 2 you can use the ‘stack’ command. i.e. go to data <select> Stack and then select columns from the options that appear. In the first box type the column identifiers for the two sets of metabolism data (c2 and c3) and then click the button ‘column of current worksheet’ and type in the column number of the column where you want the stacked data to be stored (e.g. C10). In the store subscripts box type another column name (e.g. c11) this will identify which values correspond to treatment and which to control. To get the corresponding individual IDs type C1 C1 in the ‘stack the following columns box’ this will give you the individual data in a third new column (e.g. c12). To perform the Friedman test choose Statistics – Nonparametrics – Friedman test. In the box that opens type the column identifier for the stacked metabolism data (in the above case C10). In the treatment box type the column identifier for the subscripts (C11) and in the blocks column type the column identifying the individual IDs (c12). The output is as follows. Friedman Test: metabolism versus treatment blocked by IDs S = 5.40 trtment 1 2 DF = 1 N 15 15 P = 0.020 Est Median 1.3900 1.4900 Sum of Ranks 18.0 27.0 Grand median = 1.4400 The result and P value are on the first line of the output. Step 23 Analysis of Covariance: ANCOVA Data should be organized as shown in Table 1. Go to Statistics <select> ANOVA, <select> general linear model. In the box labelled response, add the column identifier for the dependent variable (e.g. metabolism or column 2 in table 1). In the box labelled ‘model’ add the column identifiers for the treatment variable and also the covariate (ie body mass) and the treatment by covariate interaction. Eg using the data in table 1 as an example the model is specified as C3 C4 C3*C4 It is necessary to declare that body weight is a covariate in the model. To do this click on the box labelled ‘covariates’ and then type the column identifier for the covariate (in this case C3) into the box. Close this box and then click on Ok to run the analysis. The output appears as follows General Linear Model: Metabolism versus Levels Factor Levels Type fixed Levels 3 Values 1, 2, 3 Analysis of Variance for Metabolism, using Adjusted SS for Tests Source DF Seq SS Adj SS Adj MS F P Body Mass Levels Levels*Body Mass Error Total S = 0.116699 1 2 2 9 14 0.07801 1.32527 0.03604 0.12257 1.56189 0.07686 0.05598 0.03604 0.12257 R-Sq = 92.15% Term Constant Body Mass Body Mass*Levels 1 2 0.07686 0.02799 0.01802 0.01362 5.64 2.06 1.32 0.042 0.184 0.314 R-Sq(adj) = 87.79% Coef -0.3954 0.07448 SE Coef 0.7367 0.03135 T -0.54 2.38 P 0.604 0.042 -0.05786 -0.01052 0.04214 0.04386 -1.37 -0.24 0.203 0.816 The significance of the different effects is shown in the ANOVA table. In this case body mass was a significant covariate (p =0.042), and there no significant effect of treatment or interaction between body mass and treatment level was found (p>0.05). Note: see results under Part 14 where treatment levels were shown to significantly affect metabolism. The analysis of covariance here suggests that these effects on metabolism might be explained by differences in body mass between individuals and are thus not caused by the treatment. However, to perform the final analysis of these data the analysis needs to be repeated omitting the non-significant interaction effect in the model. i.e. the model should be respecified as C3 C4 This should only be done when the interaction term is NOT significant. In this analysis keep C3 as a covariate. The revised output is as follows. General Linear Model: Metabolism versus Levels Factor Levels Type fixed Levels 3 Values 1, 2, 3 Analysis of Variance for Metabolism, using Adjusted SS for Tests Source Body Mass Levels Error Total DF 1 2 11 14 S = 0.120078 Term Constant Body Mass Seq SS 0.07801 1.32527 0.15861 1.56189 Adj SS 0.06147 1.32527 0.15861 R-Sq = 89.85% Coef -0.1899 0.06559 SE Coef 0.7467 0.03177 Adj MS 0.06147 0.66264 0.01442 F 4.26 45.96 P 0.063 0.000 R-Sq(adj) = 87.08% T -0.25 2.06 P 0.804 0.063 This revised analysis excluding the interaction term shows that consistent with the data analysis in section 14 there is a significant treatment effect and an effect of body mass that just fails to reach statistical significance (p = 0.063). This emphasises the critical importance of re-running such analyses excluding non-significant interaction terms. Step 37 Correlation matrix Select statistics <select> display basic statistics and <select> correlation. In the variables box enter the column identifiers for all the predictor variables that you want to correlate together. The output is a correlation matrix. A typical matrix might be as follows for a situation where 5 organ weights are available. BAT 0.13 Skeletal muscle 0.66 0.17 0.55 0.93 1.00 0.11 0.03 1.00 0.61 Organ Liver WAT Brain Liver 1.00 0.32 1.00 WAT Brain Skeletal muscle 0.22 This table highlights that WAT and BAT are highly correlated and hence not independent predictor variables. Skeletal muscle also is quite highly correlated to the liver mass. One can proceed with the analysis ignoring these effects but one should be aware that such correlations may compromise the outcome. In this case a strong effect of WAT might emerge because of the effect of BAT on metabolism combined with the high correlation of WAT with BAT. This analysis requires the number of observations (i.e., individuals) to exceed by at least a factor of 3 the number of predictor variables included into the analysis. Hence in this situation one would have 5 predictors so for each group (i.e., treatment levels and control) one would need at least 15 individuals – and preferably many more. Interpretation of these effects depends on the complexity of the interactions. The bottom line is to diagnose an overall treatment effect controlling for these body composition variables. If there is an overall treatment effect one can establish where this occurs using the multiple range tests (TUKEY TEST and DUNCAN’S MULTIPLE RANGE TEST). Step 38: PRINCIPAL COMPONENTS ANALYSIS. To perform a principal components analysis the data for the individual organ weights need to be organised such that the organ weights are in separate columns and the organ weights for a given individual are in a single row. An example set of data is included in Appendix one. These data are 17 organ weights in grams from 30 rats. The original data were published in Selman et al (2008). To perform a principal components analysis on these data select Statistics - multivariate and principal components . In the box labelled variables type the column identifiers for the 17 organs (eg c1 – c17). In the box labelled ‘number of components to compute’ type 5. This will restrict the analysis to calculate only the first 5 components. Otherwise if this is left blank the analysis will compute n components where n is the original number of columns entered into the analysis. Click on the button labelled ‘storage’ and in the new window that opens type column identifiers for the same number of columns that you asked the program to compute. E.g. if you asked it to compute 5 components then type 5 column identifiers for example C18-c22. Click OK. Closes new window. Click OK. Runs analysis. The output looks as follows: Principal Component Analysis: Carcass, HEART, LIVER, KIDNEY, BRAIN, Brown Fat…., Eigenanalysis of the Correlation Matrix 28 cases used, 2 cases contain missing values Eigenvalue Proportion Cumulative 7.6875 0.452 0.452 3.4298 0.202 0.654 1.6138 0.095 0.749 1.0599 0.062 0.811 0.7983 0.047 0.858 0.6485 0.038 0.896 0.5503 0.032 0.929 0.3541 0.021 0.950 Eigenvalue Proportion Cumulative 0.2827 0.017 0.966 0.1803 0.011 0.977 0.1250 0.007 0.984 0.1029 0.006 0.990 0.0749 0.004 0.995 0.0463 0.003 0.997 0.0228 0.001 0.999 0.0187 0.001 1.000 Eigenvalue Proportion Cumulative 0.0044 0.000 1.000 Variable Carcass HEART LIVER KIDNEY BRAIN Brown Fat Abdominal Fat Gonadal Fat Mesenteric Fat Gonads Large Intestine (g) Small Intestine (g) Stomach Lungs Pancreas Pelage Tail PC1 0.333 0.236 0.283 0.288 0.134 -0.231 0.225 0.268 0.287 0.139 0.314 0.149 0.133 -0.019 0.276 0.328 0.229 PC2 -0.107 0.286 -0.215 0.270 0.371 -0.118 -0.333 -0.255 -0.097 -0.302 -0.014 0.058 0.318 0.372 0.120 -0.153 0.280 PC3 -0.071 0.052 0.017 0.089 0.180 0.485 0.140 0.110 0.244 -0.147 -0.098 0.574 0.339 -0.114 -0.252 -0.042 -0.267 PC4 0.141 -0.206 0.016 0.034 0.087 -0.020 -0.326 -0.279 -0.196 0.451 -0.009 0.128 0.357 -0.569 0.066 -0.055 0.164 PC5 0.077 0.378 0.100 -0.163 0.014 -0.180 -0.118 -0.280 0.204 0.502 0.209 0.066 -0.030 0.296 -0.349 -0.194 -0.317 At the top of the output the note reminds us that for this analysis to run it is necessary to have complete data for all animals. If the data for a given animal is incomplete it is excluded from the analysis. Beneath this is a table containing 17 sets of values labelled Eigenvalue, proportion and cumulative. These are the proportions of the original variance contained in the 17 computed components ordered by size. Hence the first principal component explains 45.2% of the original variation. The eigenvalue is a representation of how much better this variable is at describing the variance compared to the original variables. As there were 17 original variables they each contain 1/17th of the total variation (p = 0.0588). Since this new variable contains p = 0.452 of the variation it is 0.452/0.0588 = 7.68x better than the original variables at describing the data. Another way of thinking about the eigenvalue is that it is the number of original variables that the current variable is ‘worth’. The second principal component in this case has an eigen value of 3.43 and explains 20.2% of the variation, so the cumulative variance explained by components 1 and 2 is 65.4%. Looking at this table you can see that beyond the 4th principal component the eigenvalue falls below 1 so these variables explain less than the original variables. Moreover, the first 4 components explain together 81% of the original variation. This means that by looking at just the first 4 components retains 81% of the original information but in just 4 as opposed to 17 variables. Below the eigenvalues and variance table is a second table showing each of the original variables alongside the new principal components (PC1 to PC5). The values in this table are ‘eigenvectors’ that show the strength and direction of the association between the original variable and the new component. As you can see almost all the variables affect PC1 in a positive way and so it reflects an overall size component, while PC2 is negatively affected by all the body fat components so it is a reflection of leanness of the animals. We can use the ‘scores’ on these principal components in a general linear model (see above) in place of the original organ masses. The major advantage of this is that these principal components are by definition completely independent of each other. This makes their use in the general linear model more statistically valid. R Step 1: Normality tests The following code will perform the Anderson-Darling and the Shapiro-Wilks tests for normality on the Metabolism variable in the Table1 data frame. To perform the Anderson-Darling test using the ad.test() function, the “nortest” package must be installed. On most systems the install.packages() function can be used to install packages; otherwise, there are package installation wizards associated with all script editors. To use the “nortest” package, it must be loaded using the library() function. Anderson-Darling test install.packages("nortest") library(nortest) ad.test(Table1$Metabolism) Anderson-Darling normality test data: Table1$Metabolism A = 0.2395, p-value = 0.732 Shapiro-Wilks test shapiro.test(Table1$Metabolism) Shapiro-Wilk normality test data: Table1$Metabolism W = 0.9566, p-value = 0.6333 If the p-value is less than 0.05, then the distribution of the data differs significantly from normal. If the p-value is greater than 0.05, then the data can be considered normally distributed. Normality Q-Q plots can be made for the Metabolism variable in the Table1 data frame using the qqnorm() function. A Q-Q line that reflects what would is expected if the distribution is normal can be added using the qqline() function. Note that to make this plot with the observed values on the x-axis, datax = TRUE must be specified in both the qqnorm() and qqline() functions. qqnorm(Table1$Metabolism, datax = TRUE, xlab = "Expected Normal", ylab = "Observed Values") qqline(Table1$Metabolism, datax = TRUE) Step 3: Attempt to normalise the distribution by transforming it. New log10 or square root transformed variables can be added to the data frame using the log10() and sqrt() functions, respectively. Transformed variables (e.g. log10Metabolism or sqrtMetabolism) can be analyzed in the steps below. Table1$log10Metabolism <- log10(Table1$Metabolism) Table1$sqrtMetabolism <- sqrt(Table1$Metabolism) Alternatively data may be transformed using the BOX-COX procedure Prior to performing a Box-Cox transformation, the “MASS” library needs to be loaded using the library() function (the “MASS” package comes with the installation of R). To perform the Box-Cox transformation in R, an ANOVA or ANCOVA model needs to be specified using either the lm() or aov() functions (e.g. aov(Metabolism ~ Levels, data = Table1)). The boxcox() function calculates the log-likelihood of a sequence of Lambda values (λ) attempting to normalise the residuals of the linear model that is specified. The default of the boxcox() function is to calculate log-likelihood values for λ values between -2 and 2 at 0.1 intervals. In this example, the plateau of the log-likelihood function peaks outside these λ values; therefore, in the function below log-likelihood values between -5 and 5 at 0.1 intervals are calculated using the seq() function. The default of the boxcox() function is to plot loglikelihood values on λ values with the 95% confidence interval of λ values. In this example, the results of the boxcox() function will be placed in a list that we have arbitrarily called bc. This list contains two vectors: (1) a vector of the λ values used between -5 and 5 (this vector is called x and it can be seen with the command bc$x), and (2) a vector of all the log-likelihood values calculated from the λ values (this vector is called y and it is shown below rounded to two decimal places using the round() function). The max() function is used to determine the largest log-likelihood value. The which.max() function is used to determine the position of maximum loglikelihood value in the log-likelihood vector. In this case, the 77th value in the log-likelihood vector was the maximum value. The vector notation bc$x[] will be used to output λ value in the position corresponding to the maximum loglikelihood value; this value will be called Lambda. Finally, values transformed by the Box-Cox transformation (yi(λ)) are calculated using the following formulas: (1) if the maximum log-likelihood λ ≠ 0: yi(λ) = (yiλ – 1) / λ (shown below), (2) if the maximum log-likelihood λ = 0: yi(λ) = loge(yi) specified using the log() function in R. This transformation is then applied to the dependent variable (i.e. Metabolism) and then the ANOVA or ANCOVA model is re-run using the Box-Cox transformed dependent variable. library(MASS) bc <- boxcox(aov(Metabolism ~ Levels, data = Table1), lambda = seq(5, 5, 0.1)) bc$x [1] -5.0 -4.9 -4.8 -4.7 -4.6 -4.5 -4.4 -4.3 -4.2 -4.1 -4.0 -3.9 -3.8 -3.7 -3.6 [16] -3.5 -3.4 -3.3 -3.2 -3.1 -3.0 -2.9 -2.8 -2.7 -2.6 -2.5 -2.4 -2.3 -2.2 -2.1 [31] -2.0 -1.9 -1.8 -1.7 -1.6 -1.5 -1.4 -1.3 -1.2 -1.1 -1.0 -0.9 -0.8 -0.7 -0.6 [46] -0.5 -0.4 -0.3 -0.2 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 [61] 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0 2.1 2.2 2.3 2.4 [76] 2.5 2.6 2.7 2.8 2.9 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 [91] 4.0 4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8 4.9 5.0 round(bc$y, digits = 2) [1] -31.68 -30.83 -29.98 -29.13 -28.28 -27.44 -26.60 -25.76 -24.93 -24.10 [11] -23.27 -22.45 -21.62 -20.81 -19.99 -19.18 -18.37 -17.56 -16.76 -15.97 [21] -15.17 -14.38 -13.59 -12.81 -12.03 -11.26 -10.49 -9.72 -8.96 -8.20 [31] -7.45 -6.70 -5.96 -5.23 -4.49 -3.77 -3.05 -2.33 -1.63 -0.93 [41] -0.23 0.45 1.13 1.80 2.47 3.12 3.76 4.40 5.02 5.63 [51] 6.23 6.81 7.38 7.94 8.48 9.01 9.52 10.01 10.48 10.93 [61] 11.35 11.76 12.14 12.50 12.83 13.14 13.42 13.67 13.89 14.09 [71] 14.26 14.40 14.51 14.60 14.65 14.69 14.69 14.67 14.63 14.56 [81] 14.47 14.36 14.23 14.09 13.92 13.74 13.54 13.33 13.10 12.86 [91] 12.61 12.35 12.08 11.80 11.51 11.21 10.91 10.59 10.27 9.95 [101] 9.62 max(bc$y) [1] 14.6913 which.max(bc$y) [1] 77 Lambda <- bc$x[which.max(bc$y)] Lambda [1] 2.6 Table1$MetabolismBC <- (Table1$Metabolism^Lambda) / Lambda summary(Table1$MetabolismBC) Min. 1st Qu. 0.1157 0.5230 R Median 0.8232 Mean 3rd Qu. 0.9376 1.4030 Max. 1.7990 Step 7: Paired comparisons of parametric normally distributed data For two sample t-test Using the data presented in Table 1, the difference between the Treatments can be evaluated using the t.test() function. In this function, a two-sided test is the default comparison between the Treatments. If equal variances in the two Treatments are assumed, then the use the following code: t.test(Metabolism ~ Treatment, data = Table1, var.equal = TRUE) If equal variances in the two treatments are not assumed, then use the following code: t.test(Metabolism ~ Treatment, data = Table1, var.equal = FALSE) The output from the t.test() function where equal variances in the two treatments are not assumed is: Welch Two Sample t-test data: Metabolism by Treatment t = 5.1023, df = 7.771, p-value = 0.001014 alternative hypothesis: true difference in means is not equal to 0 95 percent confidence interval: 0.3066951 0.8173049 sample estimates: mean in group 1 mean in group 2 1.538 0.976 The t-value, df, and p-value are shown on the same line. If the p-value < 0.05 then the difference between the two Treatments is significant. In this case, Metabolism in group 1 was significantly greater than Metabolism in group 2. If you want to output the standard deviation and the standard error (standard deviation / square root of the number of samples) for both groups, then use the tapply() function. tapply(Table1$Metabolism, Table1$Treatment, sd) 1 2 0.1950954 0.2040343 tapply(Table1$Metabolism, Table1$Treatment, sd) / sqrt(tapply(Table1$Metabolism, Table1$Treatment, length)) 1 2 0.06169459 0.09124692 For Paired t-test This analysis uses the data provided in Table 2. To run this analysis, the data in Table 2 needs to be in ‘stacked format’. In the stacked format data frame, there are five columns: 1) ID, 2) Metabolism – all values from the Metabolism Control, Metabolism Treatment 1, and Metabolism Treatment 2 columns, 3) Levels - the Metabolism Control, Metabolism Treatment 1, and Metabolism Treatment 2 values are specified as Metabolism1, Metabolism2, and Metabolism3, respectively, 4) Body Mass and 5) Sex. In the stacked data, all categorical variables (ID and Sex) must be specified as factors using the as.factor() function. This paired t-test compares Metabolism between the Levels Metabolism1 and Metabolism2 (the Metabolism3 values are removed using subset not equal to “!=”). Table2stack <- read.table("Table2b.txt", header=TRUE) Table2stack$Sex <- as.factor(Table2stack$Sex) Table2stack$ID <- as.factor(Table2stack$ID) t.test(Metabolism ~ Levels, data = Table2stack, subset = Table2stack$Levels != "Metabolism3", paired = TRUE, var.equal = FALSE) Paired t-test data: Metabolism by Levels t = -3.1122, df = 14, p-value = 0.007644 alternative hypothesis: true difference in means is not equal to 0 95 percent confidence interval: -0.20269722 -0.03730278 sample estimates: mean of the differences -0.12 In this case, the difference between the Metabolism1 and Metabolism2 Levels is highly significant (i.e. P < .01). To calculate the mean within each Level, use the tapply() function. tapply(Table2stack$Metabolism, Table2stack$Levels, mean) Metabolism1 Metabolism2 Metabolism3 1.350667 1.470667 1.402000 Step 8: multiple treatment levels, parametric tests R For One way ANOVA Using the data from Table 1, to compare the Control, Treatment1, and Treatment2 Levels, use the aov() function. summary(aov(Metabolism ~ Levels, data = Table1)) Df Sum Sq Mean Sq F value Pr(>F) Levels 2 1.34181 0.67091 36.582 7.827e-06 *** Residuals 12 0.22008 0.01834 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ The p-value in this summary table is under the Pr(>F) heading. In this case, the difference between Levels is highly significant (i.e. P < .01). Repeated-measures ANOVA This analysis uses the data provided in Table 2 and compares Metabolism measured under three conditions (Levels: Metabolism1, Metabolism2, and Metabolism3). To run this analysis, the data in Table 2 needs to be in ‘stacked format’ – i.e. all the Metabolism data need to be in a single column with other four columns identifying the Levels (Metabolism1, Metabolism2, and Metabolism3), Body Mass, Sex, and individual IDs. Repeated measures ANOVA can be performed in R using either the lme() or the aov() functions. The random effect of ID is included in the lme() function by adding “random = ~1|ID” as a separate argument, whereas in the aov() function it is included by adding “+ Error(ID)” in the formula. In order to use the lme() function, the “nlme” package must be installed and loaded using the library() function. install.packages("nlme") library(nlme) anova(lme(Metabolism~Levels, random = ~1|ID, data = Table2stack)) (Intercept) Levels numDF denDF F-value p-value 1 28 445.5459 <.0001 2 28 3.7787 0.0353 summary(aov(Metabolism ~ Levels + Error(ID), data = Table2stack)) Error: ID Df Sum Sq Mean Sq F value Pr(>F) Residuals 14 2.8023 0.20017 Error: Within Df Sum Sq Mean Sq F value Pr(>F) Levels 2 0.10875 0.054376 3.7787 0.03525 * Residuals 28 0.40292 0.014390 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 The p-value in this summary table is under the Pr(>F) heading. The difference between Levels is significant (i.e. P < .05). The significance of the random effect is not provided by either the lme() or the aov() functions, but the Fvalue can be calculated using the aov() output by dividing the Mean Square of the ID effect (0.20017), by the Mean Square Residual (0.014390). Using this F-value, the p-value can be calculated based on the F distribution with degrees of freedom equal to 14 and 28 using the pf() function. In this case, the random effect of ID is highly significant (P < 0.0001). pf(0.20017 / 0.014390, df1 = 14, df2 = 28, lower.tail = FALSE) [1] 4.51458e-09 Step 10: post hoc tests For One way Anova The one-way ANOVA in step 9 suggested that Metabolism was significantly different among the Levels in Table 1. Tukey's post-hoc test can be used to determine what Levels were significantly different from each other using the TukeyHSD() function. TukeyHSD(aov(Metabolism ~ Levels, data = Table1)) Tukey multiple comparisons of means 95% family-wise confidence level Fit: aov(formula = Metabolism ~ Levels, data = Table1) $Levels diff lwr upr p adj 2-1 -0.340 -0.5685037 -0.1114963 0.0048896 3-1 -0.732 -0.9605037 -0.5034963 0.0000053 3-2 -0.392 -0.6205037 -0.1634963 0.0017004 The Levels that are being compared are given in the left-most column. Based on the p-values (p adj column), all three Levels differ significantly from each other (P < 0.005). Repeated-measures ANOVA The repeated measures ANOVA in step 9 suggested that Metabolism differed among the Metabolism1, Metabolism2, and Metabolism3 Levels. This analysis determines what Levels were significantly different from each other. The data in Table 2 needs to be in ‘stacked format’ for this analysis. Post-hoc tests following repeated measures ANOVA can be performed on lme() function objects (see Step 9 for the installation of the "nlme" package for the lme() function), but not on the aov() function objects. To proceed, the “multpcomp” package must be installed, the package must be loaded using the library() function, and the glht() function is used. library(nlme) install.packages(“multcomp”) library(multcomp) summary(glht(lme(Metabolism ~ Levels, random = ~1|ID, data = Table2stack), linfct=mcp(Levels="Tukey"))) Simultaneous Tests for General Linear Hypotheses Multiple Comparisons of Means: Tukey Contrasts Fit: lme.formula(fixed = Metabolism ~ Levels, data = Table2stack, random = ~1 | ID) Linear Hypotheses: Estimate Std. Error z value Pr(>|z|) Metabolism2 - Metabolism1 == 0 0.12000 0.04380 2.740 0.017 * Metabolism3 - Metabolism1 == 0 0.05133 0.04380 1.172 0.470 Metabolism3 - Metabolism2 == 0 -0.06867 0.04380 -1.568 0.260 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 (Adjusted p values reported -- single-step method) The Levels that are being compared are given in the left-most column. Based on the p-values (Pr(>|z|) column), Metabolism2 differed significantly from Metabolism1 (P = 0.017), but none of the other Levels differed significantly from each other (P > .05). Step 11 Power analysis To perform power analyses, the “pwr” package must be installed using the install.packages() function. To use the functions in the “pwr” package, it must be loaded using the library() function. To perform a power analysis on a two sample t-test using the pwr.t2n.test() function, you need to input the sample sizes of the two samples (i.e. n1 = 10, and n2 = 10; note that the default significance level is set to 0.05). To specify the effect size in R, the % difference that you would consider important to detect needs to be divided by the standard deviation; thus, using the same mean, % difference, and standard deviation as in the MINITAB example: d = 1.368 * 0.05 / 0.133. install.packages("pwr") library(pwr) pwr.t2n.test(n1 = 10, n2 = 10, d = 1.368 * 0.05 / 0.133) t test power calculation n1 n2 d sig.level power alternative = = = = = = 10 10 0.5142857 0.05 0.1931212 two.sided As outlined in the MINITAB example, the pwr.t.test() function can be used to calculate the sample sizes required to obtain a specified level of power (e.g. power = 0.8). pwr.t.test(power = 0.8, d = 1.368 * 0.05 / 0.133, type = "two.sample") Two-sample t test power calculation n d sig.level power alternative = = = = = 60.32651 0.5142857 0.05 0.8 two.sided NOTE: n is number in *each* group Step 13 : Two-way ANOVA Two-way ANOVA This analysis uses the data in Table 1. In order to use Type III sums-of-squares as is done in MINITAB and SPSS, the “car” package must be installed and loaded using the library() function. Two-way ANOVAs can be analyzed using the aov() function. The aov() function is nested within the Anova() function specifying that we want the analysis to use type III sums-of-squares (i.e. type = “III”). R offers a number of different ways by which factor levels can be compared (i.e. contrasted in statistical terms). Treatment contrasts are the default method in R; however, this type of contrasts is not valid for two-way ANOVAs using type III sums-of-squares. Helmert or sum contrasts are two types of contrasts that are valid for this type of ANOVA. Sum contrasts are used in the command below by specifying "contr.sum" in the function options(contrasts()). The Anova() function will output F-value, p-values, and the other values that are used to calculate them. In this example, the Levels:Sex interaction was non-significant (F2,9 = 1.3, P = 0.32), and thus it can be removed from the model. The model without the interaction suggests that there is a strong effect of Levels (F2,11 = 39.5, P < 0.0001), but that the effect of Sex is non-significant (F1,11 = 1.96, P = 0.19). install.packages(“car”) library(car) options(contrasts=c("contr.sum", "contr.poly")) Anova(aov(Metabolism ~ Levels + Sex + Levels:Sex, data = Table1), type = "III") Anova Table (Type III tests) Response: Metabolism Sum Sq Df F value Pr(>F) (Intercept) 26.6451 1 1654.4056 1.634e-11 *** Levels 1.2104 2 37.5774 4.278e-05 *** Sex 0.0333 1 2.0648 0.1846 Levels:Sex 0.0419 2 1.3000 0.3192 Residuals 0.1450 9 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 Anova(aov(Metabolism ~ Levels + Sex, data = Table1), type = "III") Anova Table (Type III tests) Response: Metabolism Sum Sq Df F value Pr(>F) (Intercept) 26.6451 1 1568.824 3.221e-13 *** Levels 1.3418 2 39.502 9.533e-06 *** Sex 0.0333 1 1.958 0.1893 Residuals 0.1868 11 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 Repeated-measures two-way ANOVA This analysis examines the effect of Levels and Sex on Metabolism including a random factor of ID. This analysis can be performed using either the lme() or the aov() functions; however, only the commands for the aov() function will be shown. The random effect of ID is included in the aov() function it is included by adding “+ Error(ID)” in the formula. For this analysis, the data in Table 2 needs to be in ‘stacked format’. The “Levels * Sex” notation suggests that both the main effects of Levels and Sex are tested in addition to their interaction. The “Levels * Sex” interaction was not significant (F2,26 = 0.06, P = 0.94), and thus, it was removed from the model. There was a significant in Metabolism among the Levels (F2,28 = 3.78, P = 0.04); however, Sex did not significantly affect Metabolism (F1,13 = 0.33, P = 0.58). summary(aov(Metabolism ~ Levels * Sex + Error(ID), data = Table2stack)) Error: ID Df Sum Sq Mean Sq F value Pr(>F) Sex 1 0.06848 0.068481 0.3256 0.578 Residuals 13 2.73383 0.210295 Error: Within Df Sum Sq Mean Sq F value Pr(>F) Levels 2 0.10875 0.054376 3.5258 0.04417 * Levels:Sex 2 0.00193 0.000967 0.0627 0.93936 Residuals 26 0.40098 0.015422 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 summary(aov(Metabolism ~ Levels + Sex + Error(ID), data = Table2stack)) Error: ID Df Sum Sq Mean Sq F value Pr(>F) Sex 1 0.06848 0.068481 0.3256 0.578 Residuals 13 2.73383 0.210295 Error: Within Df Sum Sq Mean Sq F value Pr(>F) Levels 2 0.10875 0.054376 3.7787 0.03525 * Residuals 28 0.40292 0.014390 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 Step 14: Proceed in the same way as for step 13 for Two-way ANOVA, but with additional factors added to the model formula Step 18: non parametric tests paired comparisons Mann-Whitney U/ two-sample Wilcoxon test This analysis compares Metabolism between the two Treatments in Table 1. The Mann-Whitney U test can also be called a two-sample Wilcoxon test. To determine the median of each Treatment, use the tapply() function. tapply(Table1$Metabolism, Table1$Treatment, median) 1 2 1.565 1.000 wilcox.test(Metabolism ~ Treatment, data = Table1, conf.int = TRUE) Wilcoxon rank sum test with continuity correction data: Metabolism by Treatment W = 50, p-value = 0.002647 alternative hypothesis: true location shift is not equal to 0 95 percent confidence interval: 0.2600396 0.7799804 sample estimates: difference in location 0.5543361 R gives a warning message for this analysis that it cannot calculate exact pvalues and confidence intervals because of ties. The p-value and the 95% confidence interval are corrected because there are ties amongst the Metabolism values. The p-value suggests that Metabolism is significantly different between the two Treatments (i.e. P < .05). Wilcoxon matched pairs This analysis uses the wilcox.test() function to compare the Metabolism Control and Metabolism Treatment1 columns in Table 2 assuming that these values come from the same individual (paired = TRUE). To proceed with this analysis, the Table 2 data need to be imported using: Table2 <read.table("Table2.txt", header = TRUE). wilcox.test(Table2$Control, Table2$Treatment1, paired = TRUE, conf.int = TRUE) Wilcoxon signed rank test with continuity correction data: Table2$Control and Table2$Treatment1 V = 12, p-value = 0.006945 alternative hypothesis: true location shift is not equal to 0 95 percent confidence interval: -0.20504427 -0.03504422 sample estimates: (pseudo)median -0.1072468 Again, R gives a warning message for this analysis that it cannot calculate exact p-values and confidence intervals because of ties. The p-value and the 95% confidence interval are corrected because there are ties amongst the Metabolism values. This analysis suggests that there is a significant difference between Control and Treatment1. Step 19 non parametric analysis when there are multiple treatments or levels: Kruskal-Wallis ANOVA This analysis compares Metabolism between the Treatments in Table 1 using the kruskal.test() function. The Kruskal-Wallis statistic (χ2) and the p-value adjusted for ties is given on the last line. A p-value of < .05 suggests that Metabolism is significantly different between the Treatments. kruskal.test(Metabolism ~ Treatment, data = Table1) Kruskal-Wallis rank sum test data: Metabolism by Treatment Kruskal-Wallis chi-squared = 9.4086, df = 1, p-value = 0.00216 Repeated measures Friedman test This analysis compares Metabolism between the Levels in Table2. The data in Table 2 needs to be in ‘stacked format’ and this analysis will focus on comparing Metabolism between Levels Metabolism1 and Metabolism2. The subset() function is used to omit the Metabolism3 Level and create a new data frame called Table2stacksub. The as.factor(as.character()) functions are required in this case because without performing these functions, R considers there be to zero individuals in the Metabolism3 Level and this causes an error with the Friedman Test. Table2stacksub <- subset(Table2stack, Table2stack$Levels != "Metabolism3") Table2stacksub$Levels <- as.factor(as.character(Table2stacksub$Levels)) friedman.test(Table2stacksub$Metabolism, Table2stacksub$ID, Table2stacksub$Levels) Friedman rank sum test data: Table2stacksub$Metabolism, Table2stacksub$ID and Table2stacksub$Levels Friedman chi-squared = 26.6679, df = 14, p-value = 0.02126 The significance of this test is indicated on the last line as a p-value. The pvalue is < .05 in this case suggesting that Metabolism differs significantly between the Metabolism1 and Metabolism2 Levels taking into account that these Levels were examined in the same individuals. pchisq(26.6679, df=14, lower.tail = F) [1] 0.02125833 pchisq(5.4, df=1, lower.tail = F) [1] 0.02013675 Step 23 Analysis of Covariance: ANCOVA This analysis uses the data in Table 1. In order to use Type III sums-of-squares as is done in MINITAB and SPSS, the “car” package must be installed (see Step 14 for installation of the “car” package) and loaded using the library() function. ANCOVAs can be analyzed using the aov() function. In this example, we will first run the aov() function and call the resulting model aov1. We will name the model aov1 because it will simplify the notation for the post-hoc Tukey test in step 30. Next, we will use the Anova() function on aov1 specifying that we want the analysis to use type III sumsof-squares (i.e. type = “III”). Remember that the correct contrasts need to be specified in the options() function in order for the results of Anova() to be correct (see twoway ANOVA in step 14). In this example, the Levels:Body.Mass interaction was nonsignificant (F2,9 = 1.32, P = 0.31), and thus it can be removed from the model. The ANOVA tables both including (aov1) and excluding (aov2) the Levels:Body.Mass interaction are presented. The interpretation of the analysis excluding the interaction is that there is a strong effect of Levels (F2,11 = 46.0, P < 0.0001), but that the effect of body mass is only a trend assuming a significance threshold of 0.05 (F1,11 = 4.3, P = 0.06). library(car) options(contrasts=c("contr.sum", "contr.poly")) aov1 <- aov(Metabolism ~ Body.Mass + Levels + Levels:Body.Mass, data = Table1) Anova(aov1, type = "III") Anova Table (Type III tests) Response: Metabolism Sum Sq Df F value Pr(>F) (Intercept) 0.003923 1 0.2881 0.60446 Body.Mass 0.076857 1 5.6436 0.04152 * Levels 0.055980 2 2.0553 0.18399 Body.Mass:Levels 0.036039 2 1.3231 0.31351 Residuals 0.122567 9 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 aov2 <- aov(Metabolism ~ Body.Mass + Levels, data = Table1) Anova(aov2, type = "III") Anova Table (Type III tests) Response: Metabolism Sum Sq Df F value Pr(>F) (Intercept) 0.00093 1 0.0647 0.80395 Body.Mass 0.06147 1 4.2635 0.06334 . Levels 1.32527 2 45.9567 4.561e-06 *** Residuals 0.15861 11 --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 To perform a post-hoc test on an ANCOVA, the "multcomp" package must be installed (see Step 11 for the installation of the "multcomp" package). The "multcomp" package must then be loaded using the library() function, and the glht() function is used. In the example below, the glht() function compares the Levels using a Tukey test, based on the aov2 model. library(multcomp) summary(glht(model = aov2, linfct = mcp(Levels = "Tukey"), data = Table1)) Simultaneous Tests for General Linear Hypotheses Multiple Comparisons of Means: Tukey Contrasts Fit: aov(formula = Metabolism ~ Body.Mass + Levels, data = Table1) Linear Hypotheses: Estimate Std. Error t value Pr(>|t|) 2 - 1 == 0 -0.37673 0.07800 -4.830 0.00123 ** 3 - 1 == 0 -0.72806 0.07597 -9.584 < 0.001 *** 3 - 2 == 0 -0.35133 0.07846 -4.478 0.00219 ** --Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 (Adjusted p values reported -- single-step method) Step 37 Correlation matrix Assuming that you have already imported a data frame called BodyComp into R, you can assign a new data frame called cors that only contains the organ weights that you want to compare. R uses [row, column] notation to refer to specific values within a data frame. For example, if you want to return the variable in the second row and third column of the Table1 data frame, you would type: Table1[2, 3] [1] 22.3 Because we want to have all rows from the BodyComp data frame in the cors data frame, we leave the rows position in the [row, column] notation blank. In the column position, we include all the variable names that we want to include in a vector (i.e. vectors are specified using c() notation). Finally, to display the correlation matrix using Pearson correlation coefficients, use the cor() function. The “use” argument within the cor() function allows you to deal with missing values (for more information on the “use” argument, see type: cors <- BodyComp[ , c("Liver", "WAT", "Brain", "Skeletal.Muscle", "BAT")] cor(cors, method = "pearson", use = "pairwise.complete.obs") A typical matrix might be as follows for a situation where 5 organ weights are available. BAT 0.13 Skeletal muscle 0.66 0.17 0.55 0.93 1.00 0.11 0.03 1.00 0.61 Organ Liver WAT Brain Liver 1.00 0.32 1.00 WAT Brain Skeletal muscle 0.22 This table highlights that WAT and BAT are highly correlated and hence not independent predictor variables. Skeletal muscle also is quite highly correlated to the liver mass. One can proceed with the analysis ignoring these effects but one should be aware that such correlations may compromise the outcome. In this case a strong effect of WAT might emerge because of the effect of BAT on metabolism combined with the high correlation of WAT with BAT. This analysis requires the number of observations (i.e., individuals) to exceed by at least a factor of 3 the number of predictor variables included into the analysis. Hence in this situation one would have 5 predictors so for each group (i.e., treatment levels and control) one would need at least 15 individuals – and preferably many more. Interpretation of these effects depends on the complexity of the interactions. The bottom line is to diagnose an overall treatment effect controlling for these body composition variables. If there is an overall treatment effect one can establish where this occurs using the multiple range tests (TUKEY TEST and DUNCAN’S MULTIPLE RANGE TEST). If there are large numbers of high correlations in the matrix go to 39 otherwise END. Step 38: PRINCIPAL COMPONENTS ANALYSIS. This analysis will use the BodyComp data set that was used in step 38. In order to run a PCA in R using prcomp(), individuals without complete data must be removed using the na.omit() function nested within the prcomp() function. The argument scale = TRUE is specified because the variances among the body composition variables vary considerably (the spread within each variable within the BodyComp data frame can be seen with the summary() function). The following line of code will give the proportion and cumulative proportion of the total variance explained. summary(prcomp(na.omit(BodyComp), scale = TRUE)) PC1 PC2 PC3 PC4 PC5 PC6 PC7 Standard deviation 2.7726 1.8520 1.27034 1.02950 0.89347 0.80528 0.74181 Proportion of Variance 0.4522 0.2018 0.09493 0.06235 0.04696 0.03815 0.03237 Cumulative Proportion 0.4522 0.6540 0.74889 0.81123 0.85819 0.89634 0.92871 PC8 PC9 PC10 PC11 PC12 PC13 PC14 Standard deviation 0.59503 0.53166 0.42466 0.35356 0.32082 0.2736 0.21518 Proportion of Variance 0.02083 0.01663 0.01061 0.00735 0.00605 0.0044 0.00272 Cumulative Proportion 0.94953 0.96616 0.97677 0.98412 0.99018 0.9946 0.99731 PC15 PC16 PC17 Standard deviation 0.15093 0.1366 0.06610 Proportion of Variance 0.00134 0.0011 0.00026 Cumulative Proportion 0.99865 0.9997 1.00000 Eigenvalues for all seventeen components are the square of the standard deviations given from the previous line of code and can be outputted using: prcomp(na.omit(BodyComp), scale=TRUE)$sd^2 [1] 7.687505211 3.429847979 1.613766157 1.059868403 0.798288931 0.648470172 [7] 0.550281167 0.354066373 0.282664344 0.180336227 0.125007493 0.102923245 [13] 0.074873073 0.046300902 0.022779398 0.018651206 0.004369719 To output the principle component scores associated with all seventeen organ weights (only the first four are shown) use the following line of code. The principle components show the strength of the association between the original variable and the new component. prcomp(na.omit(BodyComp), scale=TRUE) Carcass HEART LIVER KIDNEY BRAIN Brown.Fat Abdominal.Fat Gonadal.Fat Mesenteric.Fat Gonads Large.Intestine..g. Small.Intestine..g. Stomach Lungs Pancreas Pelage Tail PC1 -0.33276942 -0.23637808 -0.28312835 -0.28843254 -0.13447712 0.23057185 -0.22535429 -0.26791674 -0.28709897 -0.13919520 -0.31351304 -0.14871276 -0.13339934 0.01886844 -0.27587262 -0.32768510 -0.22887083 PC2 0.10678015 -0.28630188 0.21517741 -0.27045367 -0.37113185 0.11777104 0.33295878 0.25488352 0.09681311 0.30207318 0.01358145 -0.05786933 -0.31818402 -0.37180831 -0.12034584 0.15295340 -0.27969993 PC3 -0.07113286 0.05238709 0.01690019 0.08891807 0.18025001 0.48450263 0.14048190 0.11035956 0.24393549 -0.14657328 -0.09778719 0.57434774 0.33869275 -0.11381438 -0.25189547 -0.04247428 -0.26741094 PC4 0.140815809 -0.205892396 0.016342695 0.033666276 0.087331685 -0.020379466 -0.325546388 -0.278616530 -0.196235051 0.451415952 -0.008909068 0.128275550 0.357140179 -0.569416313 0.066332253 -0.054512252 0.163568995 The signs of the principal component scores within each principal component are arbitrary. It may be that all the scores need to be multiplied by “-1” in order to have consistent results between different statistical programs. Within a given principal component, variables with principal component scores with the same sign, have original values are correlated in the same direction with the principal component scores. For example, fifteen of the seventeen variable are correlated with PC1 in the same direction suggesting that this variable gives a general indication of body size.