Project_ShanthiIyanperumal
advertisement

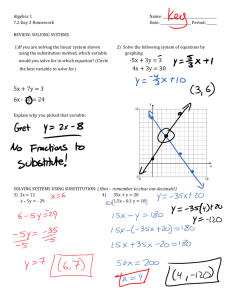
Maximum likelihood in PhylogeneticsDNA/Amino Acid Substitution Models By SHANTHI IYANPERUMAL Project for Probability and Statistics Dr. M. Partensky 4/19/04 Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 1/15 Table of Contents Maximum likelihood in Phylogenetics-.......................................................... 1 DNA/Amino Acid Substitution Models ......................................................... 1 Phylogeny ....................................................................................................................... 3 Explication of the Maximum Likelihood method ..........Error! Bookmark not defined. Calculation of likelihood of molecular sequences: ......................................................... 5 Example1: Likelihood of a single sequence with two nucleotides AC ...................... 5 Example2: Likelihood of a one branch tree between two aligned sequences ............. 5 DNA substitution Models ............................................................................................. 10 Jukes-Cantor (JC): .................................................................................................... 10 Felsenstein 1981(F81) ............................................................................................. 10 Kimura 2-parameter(K80) ....................................................................................... 10 Hasegawa-Kishino-Yano (HKY) ............................................................................ 10 Tamura-Nei (TrN):.................................................................................................... 11 Kimura 3-parameter (K3P) ..................................................................................... 11 Transition Model (TIM) .......................................................................................... 11 Transversion Model (TVM).................................................................................... 11 Symmetrical Model (SYM) .................................................................................... 11 General Time Reversible (GTR) ............................................................................. 11 Gamma Distribution (G) .......................................................................................... 11 Proportion of Invariable Sites (I) ............................................................................. 11 Amino acid Substitution Models .................................................................................. 12 Empirical substitution models................................................................................... 12 PAM matrices ........................................................................................................... 13 Dayhoff matrices ....................................................................................................... 13 JTT matrices.............................................................................................................. 13 Other empirical models ............................................................................................. 14 Blosum (Block substitution matrices) ....................................................................... 14 Poisson models.......................................................................................................... 14 Reference: ..................................................................................................................... 15 Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 2/15 Phylogeny Phylogeny is the evolution of a genetically related group of organisms. It is the study of relationships between collection of "things" (genes, proteins, organs..) that are derived from a common ancestor. Phylogeneny is used to 1. Find evolutionary ties between organisms. (Analyze changes that occured in different organisms during evolution). 2. Find or understand relationships between an ancestral sequence and it descendants. (Evolution of family of sequences) 3. Estimate time of divergence between a group of organisms that share a common ancestor. From a common ancestor sequence, two DNA sequences are diverged. Each of these two sequences start to accumulate nucleotide substitutions. The number of these mutations are used in molecular evolution analysis. One of the most striking features of life is all living organisms share highly conserved regions in proteins, particularly in proteins that are involved in information processing (transcription and translation). All share the same genetic codes. This information leads us to accept the theory that all organisms known to us have evolved from a common ancestor. Patrick Forterre coined this ancestor as LUCA (Last Universal Common Ancestor. When 2 sequences found in 2 organisms are very similar, we assume that they have derived from one ancestor. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 3/15 The sequences alignment reveal which positions are conserved from the ancestor sequence. Most phylogenetic methods assume that each position in a sequence can change independently from the other positions. General Model of DNA Substitution Maximum likelihood evaluates the probability that the choosen evolutionary model will have generated the observed sequences. Phylogenies are then inferred by finding those trees that yield the highest likelihood. The rate matrix for a general model of DNA substitution is given by . r2pC r4pG r6pT r1pA . r8pG r10pT r3pA r7pC . r12pT r5pA r9pC r11pG . Q = q(i,j) = The rows and columns are ordered A, C, G and T. The matrix gives the rate of change from nucleotide i(arranged along the rows) to nucleotide j(along the columns). For example r2pC gives the rate of change from A to C. Let P(v,s) be the transition probability matrix where p i,j(v,s) is the probability that nucleotide i changes into j over branch length v. The vector s contains the parameters of the substitution model(eg. pA, pC, pG, pT, r1,r2…). For two-state case, to calculate the probability of observing a change over a branch of length v, the following matrix calculation is performed: P (v,s) = eQv Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 4/15 Calculation of likelihood of molecular sequences: For DNA sequence comparison the model has 2 parts, the base composition and the process. The composition is just the proportion of the four nucleotides A, C, G, T. Example1: Likelihood of a single sequence with two nucleotides AC If the model is Jukes – Cantor model, which has a base composition of ¼ for each nucleotide then the likelihood will be 1/4 X 1/4 = 1/16. If the model has a composition of 40%a and 10%c the likelihood of the sequence will be 0.4 x 0.1=0.04 If we take the 16 possible nucleotide combinations and calculate the sum of all of them the sum of those likelihoods is 1. For any model, the sum of the likelihoods of all the different data possibilities should be 1. Example2: Likelihood of a one branch tree between two aligned sequences Sequence 1 CCAT Sequence 1 CCGT The other part of the model, the process part is needed if we have more than one sequence related by a tree. The process might be described by sentences, or by a matrix of numbers, describing how the nucleotides change from one to another. Let the composition part of the model be denoted by = [0.1, 0.4, 0.2, 0.3]. The order of the bases is A, C, G, and T. There are 16 possible changes from one nucleotide to the other. The changes can be represented as a 4 X 4 matrix. A P= 0.976 0.002 0.003 C 0.01 0.983 0.01 G T 0.007 0.007 0.005 0.01 0.979 0.007 A C G Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 5/15 0.002 0.013 0.005 0.979 T Likelihood of going from sequence1 to sequence 2 is: = c Pc-c c Pc-c a Pa-g t Pt-t = 0.4 * 0.983 * 0.4 * 0.983 * 0.1* 0.007 * 0.3 * 0..979 = 0.0000300 In the above example we did not consider branch lengths. Intuitively for short branch lengths the probability of a base change is low and for long branch lengths it is high. Let’s assume the matrix we have chosen describes a branch with a Certain Evolutionary Distance (CED). The likelihood we calculated was for 1 CED. The likelihood for the same alignment for 2 CED units is found by multiplying matrix P by itself. P2 = 0.953 0.005 0.007 0.005 0.02 0.013 0.966 0.01 0.02 0.959 0.026 0.01 0.015 0.02 0.015 0.959 A C G T Likelihood for 2 CED units is: = c Pc-c c Pc-c a Pa-g t Pt-t = 0.4 * 0.966 * 0.4*0.966 * 0.1* 0.013 * 0.3 * 0..959 = 0.0000559 As the branch length increases the values on the diagonal decrease and the other values increase because change becomes more likely than being the same. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 6/15 The table lists the likelihoods for increasing branch lengths. Branch length (CED units) 1 2 3 10 15 20 30 Likelihood 0.0000300 0.0000559 0.0000782 0.000162 0.000177 0.000175 0.000152 The likelihood rises to a maximum somewhere between 15 and 20 ced units. Example 3 : Likelihood of a tree with four taxa Assume that we have the aligned nucleotide sequences for four taxa: The possible trees are We want to evauate the likelihood of the unrooted tree represented by the nucleotides of site j in the sequence and shown below: Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 7/15 Since most of the models currently used are time-reversible, the likelihood of the tree is generally independent of the position of the root. Therefore it is convenient to root the tree at an arbitrary internal node as done in the Fig. below, Under the assumption that nucleotide sites evolve independently (the Markovian model of evolution), we can calculate the likelihood for each site separately and combine the likelihood into a total value towards the end. To calculate the likelihood for site j, we have to consider all the possible scenarios by which the nucleotides present at the tips of the tree could have evolved. So the likelihood for a particular site is the summation of the probablilities of every possible reconstruction of ancestral states, given some model of base substitution. So in this specific case all possible nucleotides A, G, C, and T occupying nodes (5) and (6), or 4 x 4 = 16 possibilities : Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 8/15 In the case of protein sequences each site may occupy 20 states (that of the 20 amino acids) and thus 400 possibilities have to be considered. Since any one of these scenarios could have led to the nucleotide configuration at the tip of the tree, we must calculate the probability of each and sum them to obtain the total probability for each site j. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 9/15 The likelihood for the full tree then is product of the likelihood at each site. L= L(1) x L(2) ..... x L(N) Since the individual likelihoods are extremely small numbers it is convenient to sum the log likelihoods at each site and report the likelihood of the entire tree as the log likelihood. N ln L= ln L(1) + ln L(2) ..... + ln L(N) = SUM ln L(j) j=1 DNA substitution Models The use of maximum likelihood (ML) algorithms in developing phylogenetic hypotheses requires a model of evolution. The frequently used General Time Reversible (GTR) family of nested models encompasses 64 models with different combinations of parameters for DNA site substitution. The models are listed here from the least complex to the most parameter rich. Jukes-Cantor (JC): Equal base frequencies, all substitutions equally likely . 1 level of nesting. Felsenstein 1981(F81) Variable base frequencies, all substitutions equally likely . 1 level of nesting. Kimura 2-parameter(K80) Equal base frequencies, variable transition and transversion frequencies . 2 levels of nesting. Hasegawa-Kishino-Yano (HKY) Variable base frequencies, variable transition and transversion frequencies. 2 levels of nesting. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 10/15 Tamura-Nei (TrN): Variable base frequencies, equal transversion frequencies, variable transition frequencies Kimura 3-parameter (K3P) Variable base frequencies, equal transition frequencies, variable transversion frequencies Transition Model (TIM) Variable base frequencies, variable transitions, transversions equal Transversion Model (TVM) Variable base frequencies, variable transversions, transitions equal Symmetrical Model (SYM) Equal base frequencies, symmetrical substitution matrix (A to T = T to A) General Time Reversible (GTR) Variable base frequencies, symmetrical substitution matrix . 6 levels of nesting . In addition to models describing the rates of change from one nucleotide to another, there are models to describe rate variation among sites in a sequence. The following are the two most commonly used models. Gamma Distribution (G) Rate heterogeneity can be accommodated by specifying that the rate of evolution across different sites and is distributed according to a gamma distribution. A simpler way of accounting for rate heterogeneity is to specify that a fixed proportion of sites are invariant i.e. have zero rate of evolution. Proportion of Invariable Sites (I) Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 11/15 Extent of static, unchanging sites in a dataset . Amino acid Substitution Models The divergence among sequences can be modeled with a mutation matrix. The matrix, denoted by M, describes the probabilities of amino acid mutations for a given period of evolution. This corresponds to a model of evolution in which amino acids mutate randomly and independently from one another but according to some predefined probabilities depending on the amino acid itself. This is a Markovian model of evolution and while simple, it is one of the best models. Intrinsic properties of amino acids, like hydrophobicity, size, charge, etc. can be modeled by appropriate mutation matrices. Dependencies which relate one amino acid characteristic to the characteristics of its neighbors are not possible to model through this mechanism. Amino acids appear in nature with different frequencies. These frequencies are denoted by fi and correspond to the steady state of the Markov process defined by the matrix M., i.e., the vector f is any of the columns of or the eigenvector of M whose corresponding eigenvalue is 1 (Mf=f). This model of evolution is symmetric, i.e., the probability of having an i which mutates to a j is the same as starting with a j which mutates into an i. The following is a list of amino acid substitution models which use matrices. Empirical substitution models In contrast to DNA substitution models, amino acid replacement models have concentrated on the empirical approach. Dayhoff and coworkers developed a model of protein evolution which resulted in the development of a set of widely used replacement matrices. In the Dayhoff approach, replacement rates are derived from alignments of protein sequences that are at least 85% identical; this constraint ensures that the likelihood of a particular mutation being the result of a set of successive mutations is low. One of the main uses of the Dayhoff matrices has been in database search methods where, for example, the matrices P(0.5), P(1) and P(2.5) (known as the PAM50, PAM100 and PAM250 matrices) are used to assess the significance of proposed matches between target and database sequences. However, the implicit rate matrix has been used for phylogenetic applications. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 12/15 PAM matrices In the definition of mutation the matrix M implies certain amount of mutation (measured in PAM units). A 1-PAM mutation matrix describes an amount of evolution which will change, on the average, 1% of the amino acids. In mathematical terms this is expressed as a matrix M such that The diagonal elements of M are the probabilities that a given amino acid does not change, so (1-Mii) is the probability of mutating away from i. If we have a probability or frequency vector p, the product Mp gives the probability vector or the expected frequency of p after an evolution equivalent to 1-PAM unit. Or, if we start with amino acid i (a probability vector which contains a 1 in position i and 0s in all others) M*i (the ith column of M) is the corresponding probability vector after one unit of random evolution. Similarly, after k units of evolution (what is called k-PAM evolution) a frequency vector p will be changed into the frequency vector Mk p. Notice that chronological time is not linearly dependent on PAM distance. Evolution rates may be very different for different species and different proteins. Dayhoff matrices Dayhoff presented a method for estimating the matrix M from the observation of 1572 accepted mutations between 34 superfamilies of closely related sequences. Their method was pioneering in the field. A Dayhoff matrix is computed from a 250-PAM mutation matrix, used for the standard dynamic programming method of sequence alignment. The Dayhoff matrix entries are related to M250 by . JTT matrices Jones et al. and Gonnett et al. have used much the same methodology as Dayhoff, but with modern databases. The Jones et al. model has been implemented for phylogenetic analyses with some success. Jones et al. have also calculated an amino acid replacement matrix specifically for membrane spanning segments. This matrix has remarkably different values from the Dayhoff matrices, which are known to be biased toward watersoluble globular proteins. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 13/15 Other empirical models Adachi and Hasegawa have implemented a general reversible Markov model of amino acid replacement that uses a matrix derived from the inferred replacements in mitochondrial proteins of 20 vertebrate species. The authors show that this model performs better than others when dealing with mitochondrial protein phylogeny. Blosum (Block substitution matrices) Henikoff and Henikoff have used local, ungapped alignments of distantly related sequences to derive the BLOSUM series of matrices. Matrices of this series are identified by a number after the matrix (e.g. BLOSUM50), which refers to the minimum percentage identity of the blocks of multiple aligned amino acids used to construct the matrix. These matrices are directly calculated without extrapolations, and are analogous to transition probability matrices P(T) for different values of T, estimated without reference to any rate matrix Q. The BLOSUM matrices often perform better than PAM matrices for local similarity searches, but have not been widely used in phylogenetics. Poisson models A simple, non-empirical model of amino acid replacement was proposed by Nei(1987) This model implements a Poisson distribution, and gives accurate estimates of the number of amino acid replacements when species are closely related. Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 14/15 Reference: 1. Phylogeny Estimation and Hypothesis Testing Using Maximum Likelihood by Huelsenbeck J. and Crandall K. 2. http://workshop.molecularevolution.org/resources/models/codonmodels.php 3. www.cs.technion.ac.il/~dang/courseCB/lecture13.pps 4. http://www.biology.usu.edu/biol6750/Lecture_15.htm 5. bio.wayne.edu/mf/teaching/BIO6060_Maximumlikelihood2 6. http://www.nmu.edu/biology/Lindsay/teaching/BI315/phylo/phylo_DNAmodels.html 7. http://www.icp.ucl.ac.be/~opperd/private/max_likeli.html 8.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Ab stract&list_uids=8642615 9. http://stat-www.berkeley.edu/users/terry/PMMB/Workshop2000/Lab3/phylo.pdf 10.http://www.biology.duke.edu/rausher/phylo1.pdf 11. Maximum Likelihood: Phylogeny Estimation- Neelima Lingareddy Maximum Likelihood-DNA Substitution Models – Shanthi Iyanperumal 15/15