Journal of Hepatology 42 (2005) 860–869
www.elsevier.com/locate/jhep
Global gene repression in hepatocellular carcinoma and fetal liver,
and suppression of dudulin-2 mRNA as a possible marker
for the cirrhosis-to-tumor transition
Cédric Coulouarn1, Céline Derambure1, Grégory Lefebvre1, Romain Daveau1, Martine Hiron1,
Michel Scotte1,2, Arnaud François3, Maryvonne Daveau1, Jean-Philippe Salier1,*
1
Inserm Unité 519 and Institut Fédératif de Recherches Multidisciplinaires sur les Peptides, Faculté de Médecine-Pharmacie,
22 Bvd Gambetta, 76183 Rouen cedex, France
2
Service de Chirurgie Générale et Digestive, Centre Hospitalier Universitaire, Rouen, France
3
Départment de Pathologie, Centre Hospitalier Universitaire, Rouen, France
Background/Aims: Whether the transcriptional reprogramming process induced by hepatocellular carcinoma
recapitulates that of the developing liver is at present unclear.
Methods: With a complete coverage of the liver transcriptome by microarray using adult livers as controls, we
searched for similarities and differences in mRNA abundances between hepatocellular carcinoma nodules and fetal
livers taken before (early) or after (late) the 22–24th week of gestation. Changes in some mRNA levels were studied in
further liver samples by quantitative RT-PCR.
Results: Altered gene expression in hepatocellular carcinoma mostly results in down-regulated mRNAs which largely
overlap with those repressed in the late fetal liver. Different frequencies of transcription factor binding sites in the
down-regulated genes vs control genes as well as changes in abundance of mRNAs for relevant transcription factors
point to a transcriptional repression. The down-regulated mRNAs code for proteins involved in (i) transcription and
translation, (ii) specific functions of the differentiated hepatocyte or (iii) activation of proliferation and apoptosis.
Conclusions: Apoptosis limitation is likely to predominate over active proliferation during liver development and
hepatocellular carcinoma. Repression of the apoptosis-associated dudulin-2 mRNA points to a potential marker for the
transition from a carcinoma-free to carcinoma-associated cirrhosis.
q 2005 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Keywords: Cancer; Development; Dudulin-2; Fetal liver; Hepatocyte; Microarray; Protein function; Transcription
factor; Transcriptome
1. Introduction
Hepatocellular carcinoma (HCC) is a primary liver
cancer, the main causes of which are hepatitis B or C
virus infection, alcohol abuse or aflatoxin B1 intoxication.
In most instances, HCC develops in the setting of chronic
hepatitis or cirrhosis [1]. Numerous HCC-associated
epigenomic alterations result in a dysregulated expression
Received 29 November 2004; received in revised form 27 January 2005;
accepted 28 January 2005; available online 11 April 2005
* Corresponding author. Tel.: C33 235 14 84 59; fax: C33 235 14 85 41.
E-mail address: jean-philippe.salier@univ-rouen.fr (J.-P. Salier).
of genes and proteins [1]. Liver transcriptome analysis by
microarray has resulted in the identification of hundreds of
genes with an aberrant under- or over-expression in HCC as
compared to the surrounding cirrhotic tissue [2–7].
However, when cumulated, many of these data appear to
be blurred or sometimes contradictory, and an overall
picture of altered gene regulation remains elusive [1,6,8].
Liver development entails the ordered activity of
transcription factors which are mostly liver-enriched
transcription factors (LETF) [9–11]. These LETFs orchestrate the up- or down-regulation of numerous target genes in
the fetal hepatocyte, a cell which must simultaneously adapt
to changes in body metabolism, escape apoptosis
0168-8278/$30.00 q 2005 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
doi:10.1016/j.jhep.2005.01.027
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
and proliferate [9–11]. Among such target genes, the
a-fetoprotein (AFP) gene is highly expressed in fetal as
opposed to adult liver and its re-expression is frequently
observed in HCC [12]. Other genes have been shown to
exhibit a similar up-regulation in fetal liver (FL) and HCC
as compared to adult liver [13–16]. In fact, it has been
suspected that the transcriptional reprogramming induced in
HCC could mimic that of the developing liver [16,17]. Yet,
genome-wide studies during liver development have
focused on a relatively small number of regulated genes
[18–21] and the few transcriptome studies that have
compared FL vs HCC provided limited information on a
possible gene overlap [16,17]. Hence, one still lacks
complete data that would point to the global similarities
and differences of gene expression in FL vs HCC. With a
microarray covering every gene expressed in fetal, adult or
tumorous liver [22], we have now identified a general trend
to gene under-expression in FL as well as HCC, as
compared to normal adult liver. This appears to be
controlled, at least partly, by a limited number of [LETF/
binding site] combinations. The mRNAs repressed in FL
and HCC are prominantly involved in transcription/
translation, cell proliferation/apoptosis, or differentiated
hepatocyte metabolism, and one of them provides a
prognostic marker for the cirrhosis-to-HCC transition.
861
that obtained from other, cumulated analyses [6]. The Genesis tool was
used for data analysis by clustering [24]. Data from the Gene Ontology
Consortium [25] were used to retrieve information on protein functions.
Quantitative Reverse Transcription PCR (q-RT-PCR) of mRNAs was done
as described [22] and the primers are listed as a supplementary Table S1 on
the journal web site (doi:10.1016/j.jhep.2005.01.027). Determination of
dudulin-2 mRNA level was done with an Assays-on-Demand kit
(ref 4331182) and a Taqman 7700 equipment from ABI. Every mRNA
level were normalized with the 18S mRNA level.
2.3. Computerized search for LETF binding sites
and comparison of occurrence in classes
Gene promoter sequences were retrieved with the EZ-Retrieve program
[26]. Control promoters from genes that appeared not to be regulated in this
study were chosen on the sole basis of promoter sequence availability. For
every gene, the first 5 kb of sequence upstream of the transcription start site
were used for a search of potential binding sites for any transcription factor
with the TRANSFAC library [27] and the vertebrate option of the
TFSEARCH program (http://www.cbrc.jp/research/db/TFSEARCH.html).
When a potential binding site was thus identified, this site was retained for
further analysis provided it was present in at least 30% of the promoters of
at least one of the two classes of regulated or control genes. Differences in
occurrence of this binding site between classes were evaluated with a nonparametric Wilcoxon’s test. For the binding sites listed in Section 3, the
threshold for a similarity between a consensus and an actual sequence
within a promoter was set at 80% (CDP, CRE, E47, GATA, HSF1, Ik-2,
XBP-1) or 90% (AP-1, C/EBP, HSF2, NF-kB).
3. Results
2. Patients and methods
3.1. Comparison of mRNA levels in FL, adult liver
and HCC by microarray
2.1. Human subjects and RNA sources
Liver fragments were obtained under strict anonymity from the
digestive surgery unit of Charles Nicolle Hospital (Rouen, France). The
clinical data are provided in Table 1. A fragment of a cancerous nodule as
well as distant cirrhotic tissue were taken whenever an HCC resection was
performed. HCC-free cirrhotic liver was obtained by surgical biopsy for
histological diagnosis in patients who were operated on for various, nonliver-related pathologies but presented cirrhosis suspicion at surgery.
Control human liver was obtained mostly in patients operated on for benign
liver tumor or liver metastasis of a non-hepatic cancer, in which cases the
tissue was taken away from the tumor. According to the current French
rules and ethical guidelines, neither an informed consent nor an advice from
an ethical committee are requested prior to analysis of RNA in resected
tissues that would otherwise be disposed off. Histopathology was carried
out by a trained pathologist. Tissue storage, culture of hepatoma cell lines
and RNA extraction were done as described [22]. Following crude liver cell
dissociation by collagenase treatment, hepatocytes and non-parenchymal
cells were separated by centrifugation, as described [23]. Two sets of early
or late FL RNAs (a pool of several livers covering the 15–24th week of
gestation or another pool of 38 livers covering the 22–40th week of
gestation, respectively) were purchased from Clontech.
2.2. Transcriptome analysis
Our set of human cDNA probes dubbed Liverpool that is tailored to a
complete coverage of the human liver transcriptome (ca. 10,000 genes), the
associated LiverTools database, as well as the procedures from array
preparation to final data handling have all been detailed [22]. In this study,
every RNA sample was subjected to three rounds of hybridization and the
resulting, normalized values were used for a selection of regulated mRNAs,
i.e. whose abundance varies significantly (P!0.05) between samples [22].
Such triplicates result in a false discovery rate that is below 10% of the total
number of regulated mRNAs (not detailed). This figure is consistent with
We analyzed liver samples with a microarray that
provides a complete coverage of the liver transcriptome
[22]. The developmental stage was taken into account with
two samples of FL RNAs pooled from either 15–24-weekold (early FL) or 22–40-week-old fetuses (late FL). We also
used RNAs from nine HCCs and four control adult livers
(HCC1–HCC9 and C1–C4 in Table 1). We selected every
mRNA whose level was significantly different in early FL
and/or late FL and/or at least three HCC samples, as
compared to the mean level found in controls. This resulted
in a selection of 1436 mRNAs, as summarized in Fig. 1A.
The most immediate observation is the very high fraction of
down-regulated mRNAs found in HCCs (83.9% of 704
mRNAs). In fact, this feature was obtained whatever the
number of HCC samples chosen for the cut-off noted above
(data not shown). Moreover, a predominating downregulation of mRNAs levels was also observed in early
FL (63.0% of 466 mRNAs) and late FL (60.0% of 647
mRNAs). We then used a principal component analysis
(PCA) whereby the samples were gathered on the basis of
similarity in mRNA abundance pattern. The PCA presented
in Fig. 2 immediately pointed to a shared location of the four
control livers within a narrow three-dimensional space.
Moreover, the early FL was located apart from all other
samples whereas the late FL and the nine HCCs shared the
same broad area (of note, no particular location or clustering
862
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
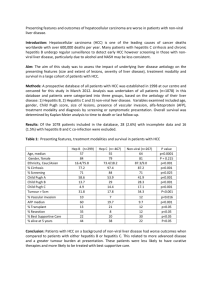
Table 1
Clinical data in patients with HCC or HCC-free cirrhosis, and control patients
Patient
Sex
Age
Pathology
Etiology
Tumor
gradea
Number of
nodules
Vascular
invasion
HCC1
HCC2
HCC3
HCC4
HCC5
HCC6
HCC7
HCC8
HCC9
M
M
M
M
M
M
M
M
M
60
63
69
49
73
72
61
45
49
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
4
2
3
1
2
3
3
2
1
O6
1
1
1
1
1
1
1
3
Yes
No
No
No
No
Yes
No
No
No
HCC10
HCC11
HCC12
HCC13
HCC14
HCC15
HCC16
HCC17
HCC18
HCC19
HCC20
HCC21
HCC22
HCC23
HCC24
HCC25
HCC26
HCC27
CIR1
CIR2
CIR3
CIR4
CIR5
CIR6
C1
C2
C3
C4
C5
C6
C7
C8
C9
C10
C11
C12
C13
M
M
M
M
F
M
F
M
M
M
M
M
M
M
M
M
M
F
M
F
F
F
M
F
M
M
M
F
F
M
F
F
F
F
F
F
M
70
65
71
65
73
68
73
66
52
72
68
78
56
68
70
82
49
42
65
55
50
50
45
48
62
76
74
94
71
72
68
42
62
53
30
45
57
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
HCC
CIR
CIR
CIR
CIR
CIR
CIR
Meta. (colon)b
Meta. (colon)
Meta. (colon)
Meta. (breast)
Hemangioma
Meta. (colon)
Meta. (breast)
HCCc
CHGc
Hemangioma
FNHc
Adenoma
Hemangioma
Hemochromat
HCV
HCV
ALC
ALC
ALC
HCVCALC
HCVCALC
HBVCHCVC
ALC
HCV
HCV
HCV
HCV
HCV
ALC
ALC
ALC
ALC
ALC
ALC
ALC
ALC
ALC
ALC
HBV
HBVCALC
None of above
HCV
HCV
ALC
ALC
ALC
HCV
1
2
2
3
3
1
2
2
2
2
2
3
3
3
3
2
3
2
1
1
2
2
1
2
1
1
3
3
7
1
1
1
1
1
1
1
No
No
No
No
Yes
Yes
No
No
Yes
Yes
Yes
No
Yes
No
No
Yes
Yes
No
HCC, patient with hepatocellular carcinoma; CIR, patient with an HCC-free cirrhosis; C, control patient; HBV, hepatitis B virus infection; HCV, hepatitis C
virus infection; ALC, alcohol abuse.
a
Differentiation grade [46].
b
Whenever a control liver was resected for a hepatic metastasis in the follow-up of a non-hepatic cancer, the tumor origin is noted in brackets.
c
Histologically normal liver taken away from (i) an HCC nodule of unknown etiology developed in a non-cirrhotic liver (HCC), or (ii) a cholangiocarcinoma
(CHG), or (iii) a focal nodular hyperplasia (FNH).
of any HCC sample related to etiology, grade, tumor size or
vascular invasion could be found here). This suggested that
the similarity of gene expression between FL and HCC was
closest with late FL.
We next focused on the mRNAs whose abundance was
found to be different in FL and HCC samples vs control
adult livers. The data are presented in Fig. 1B. Remarkably,
the very limited fraction of mRNAs with an opposite
regulation in late FL vs HCCs (4.5% of 332 mRNAs)
contrasted with a relatively high fraction of
opposite regulations in early FL vs HCCs (24.6% of 126
mRNAs). This again indicated a global similarity of gene
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
mRNAs regulated in :
late FL
n=647
early FL
n=466
B
HCC
n=704
mRNAs regulated in :
HCC & early FL
HCC & early FL
& late FL
n=33
n=93
HCC & late FL
n=299
332
126
up-regulation
opposite
regulation
down-regulation
Fig. 1. Similarities and differences in mRNA levels regulated in early,
late FL and/or HCCs vs controls. The diagrams depict the numbers of
mRNAs whose levels were found to be up- or down-regulated in a given
condition vs controls (A) or in a similar or opposite direction between
conditions (B). A given mRNA was included whenever it was found to
be regulated in at least the early or late FL or at least three out of nine
HCC samples as compared to its average value in four control adult
livers, as determined by microarray. In A, the total number of mRNAs
is only 1436, for redundancy between subsets.
down-regulation in late FL and HCC. A bi-dimensional
hierarchical clustering based onto the subset of mRNAs
included in Fig. 1B is presented in Fig. 3. Again, the late FL
clustered with a subset of three HCCs, whereas the early FL
was apart from all other samples, alike what was
observed above by PCA with a much higher number of
mRNAs. The data in Fig. 3 further demonstrated that all
nine HCCs and late FL shared a strong down-regulation
of numerous mRNA levels (Section 1, 65.6% of all mRNAs
and Section 3, 24.6%) and an up-regulation of a very
limited number of other mRNA levels (Section 2, 9.8% of
all mRNAs), as compared to the control livers. In contrast,
the early FL exhibited quite a limited number of downregulated mRNAs (Section 3). Overall, these data
again demonstrated that (i) altered gene expression in
HCC mostly results in a decreased abundance of the
corresponding mRNAs and (ii) this under-expression is
also found in the late FL, i.e. after the 22–24th week of
gestation.
early FL
C4
C3
C2
C1
HCC2
HCC3
late FL
HCC1
HCC5
HCC7
HCC4
HCC9
HCC8
HCC6
A
863
I
y
z
II
6
x
8
4
4
9
1
late
Control
1
3 2
III
2
3
5
7
Fold
induction
early
Fetal liver
HCC
Fig. 2. Clustering of liver sources by PCA. The 1436 mRNA levels
found to be significantly up- or down-regulated in at least early or late
FL (open square) or three HCCs (closed triangle 1–9) as compared to
their mean level in control livers (open dot 1–4), i.e. the 1436 mRNAs in
Fig. 1A, were used for PCA. The axes depict the first three variance
components of the mRNAs included in the analysis, which together
represent 84.9% of the total variance.
x3
1:1
x3
Fold
repression
Fig. 3. Clustering of samples or mRNAs by bi-dimensional hierarchical
clustering. The 425 mRNAs found to be significantly regulated in at
least early FL or late FL, as well as in at least three HCC samples as
compared to their mean level in controls, i.e. the 425 mRNAs in Fig. 1B,
were used for bi-dimensional hierarchical clustering (average linkage
option). The samples are clustered horizontally and the mRNAs are
clustered vertically (the gene symbols are omitted). In every sample a
change (fold) in a given mRNA level relative to its mean level in the four
control samples is shown as a colored bar of variable intensity (scale at
the bottom). Subsets I or II contain mRNAs that are, respectively,
down- or up-regulated in late FL and HCCs. Subset III contains
mRNAs that are down-regulated in early FL, late FL and HCCs.
864
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
3.2. LETF binding sites in the genes
for down-regulated mRNAs
Table 2
Relative abundance of a given LETF-encoding mRNA in FLs or HCCs
vs controls
We next investigated whether given LETF binding sites
in the genes for some of the mRNAs above could account
for the similar down-regulation seen in late FL and HCC.
From the subset of mRNAs down-regulated in late FL and
HCCs (i.e. most of the 332 mRNAs in Fig. 1B), we first
selected every gene for which at least 5 kb of promoter
sequence were available, which resulted in 21 such genes,
here below designated the down-regulated genes. As a
control, we screened a set of genes with an expression at
least in liver [22] and without any particular regulation of
the corresponding mRNA in this study (i.e. not included in
Fig. 1). We randomly retained 48 such genes for which a
promoter sequence was available. A computerized search
for any potential binding site disclosed a significant
difference (P!0.05) in the number of binding sites for 11
distinct LETFs between the two classes of 21 downregulated vs 48 control genes. This included an increased
frequency of binding sites for four LETFs (AP-1, C/EBP,
NF-kB, XBP) along with a decreased frequency of binding
sites for seven other LETFs (CDP, CRE, E47, GATA,
HSF1, HSF2, Ik-2) in the down-regulated genes. These data
indicate that the global mRNA down-regulation seen in late
FL and HCC is, at least partly, accounted for by an increased
or decreased probability of LETF binding to the corresponding gene promoters.
This mechanism in turn implies a change in abundance
of the cognate LETFs during the events under study.
Therefore, we also examined whether the abundances of
the corresponding LETF-encoding mRNAs were modified
in our FL and/or HCC samples vs adult livers. Although
many differences in abundance of such mRNAs between
these groups did not reach statistical significance, the
corresponding hierarchical clustering, shown as a supplementary Fig. S1, pointed to changes in mRNA
abundances, mostly for some members of the AP-1,
ATF, C/EBP, GATA and XBP families. Again, a
similarity of the late FL and HCCs was observed whereas
the early FL was located apart. Moreover, Table 2
presents significant changes in the abundance of given
LETF mRNAs. When a significant change was observed
in either HCC or late FL, a similar trend was
concomitantly found in the other condition. Remarkably,
we noted opposite regulations for related LETF mRNAs,
as examplified with (i) C/EBPa and -g at the C/EBP sites,
and (ii) c-Jun, JunB, ATF-3 and ATF-7 at the AP-1 sites.
This dynamic is consistent with a change of activity of the
target promoters [28,29]. Collectively, our findings
indicate that the condition-associated abundance of given
LETFs along with the specific frequency of their binding
sites onto some liver-expressed genes can explain, at least
partly, the similar down-regulation of some hepatic
mRNAs observed in late FL and HCC.
LETF binding site
a
AP-1 (C)
C/EBP (C)
NF-kB (C)
XBP (C)
CRE (K)
E47 (K)
GATA (K)
Ik-2 (K)
LETF
LETF mRNA
Early FL/C
c-Jun
JunB
JunD
c-Fos
C/EBPa
C/EBPb
C/EBPg
C/EBPd
p105
XBP-1
ATF-1
ATF-2
ATF-3
ATF-4
ATF-5
ATF-6
ATF-7
E47
GATA-4
GATA-6
Ikaros
b
0.54
0.70
0.97
0.89
1.00
0.82
0.87
0.72
0.90
0.98
1.04
0.85
1.12
1.38
0.80
0.69
2.530.70
1.13
0.65
1.42
Late FL/C
HCC/C
0.47
2.00
1.29
0.84
0.89
1.28
2.05
0.95
1.45
2.15
0.89
0.85
2.451.18
0.88
1.29
0.75
0.410.83
1.82
0.41-
0.301.461.19
0.86
0.641.13
1.421.02
1.24
1.660.77
1.03
1.651.19
1.03
0.73
0.290.84
1.13
1.24
0.36-
a
A sequence with similarity to this binding site exhibits a significantly
high (C) or low frequency (K) in the promoters of genes whose mRNAs
are down-regulated in both late FL and HCC vs control promoters (see
Section 3). Accession numbers for binding sites in the TRANSFAC data
library: AP-1, M00173; C/EBP, M00116; CRE, M00040; E47, M00071;
GATA, M00075; Ik-2, M00087; NF-kB, M00052; XBP, M00251.
b
Value presented as a ratio: normalized abundance of mRNA in the
indicated sample/average of normalized abundances in four control adult
livers (C). In the HCC/C column, the mean ratio for nine samples is
given. A star indicates a significant difference (P!0.05) between FL vs
controls (estimated as in 22) or between HCCs vs controls (one-way
analysis of variance).
3.3. Functionally defined subsets of down-regulated
mRNAs found in late FL and HCC
We searched whether functionally defined mRNAs that
were similarly down-regulated in late FL and HCC could
help identify some cellular events that are shared in these
two conditions. Within the subset of 332 regulated mRNAs
in Fig. 1B, 158 mRNAs corresponded to proteins with
defined functions, as exemplified in Table 3 and detailed in
our supplementary Table S2 on line. Remarkably, we
identified three prominent functional subsets in which most
or all mRNAs were down-regulated. They cover transcription and translation, or cell proliferation and apoptosis, or
they are hallmarks of the differentiated hepatocyte, as
follows.
Actors of the transcriptional machinery included, for
instance: (i) member 2 of SWI/SNF subfamily c, a
modifier of chromatin structure, (ii) Fos-like antigen-1
and -2 that participate in AP-1 formation and the
MKP-1-like protein tyrosine phosphatase that controls
AP-1 activation, (iii) the LETF STAT-3 and its inhibitor
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
865
Table 3
Functional classification of mRNAs with a regulated abundance in HCCs and late FL vs control liversa
Transcription/translation
Transcription (up-regulatedb)
FOS-like antigen-1
MKP-1 like protein Tyr phosphatase
Transcription (down-regulatedb)
FOS-like antigen 2
SWI/SNF subfamily c, member 2
Cell death-regulatory protein GRIM19
STAT3
Translation (down-regulatedb)
Ribosomal protein L29
Ribosomal protein S3A
Ribosomal protein S18
Eukaryotic transl. elongation factor 1a1
Ribosomal protein L13a
Deoxyhypusine synthase
DEAD box polypeptide 6
Ribosomal protein S26
Cell proliferation/apoptosis
Cell proliferation (down-regulatedb)
Interleukin 6 receptor
Peroxiredoxin 1
DEAD box polypeptide 6
Apoptosis (down-regulatedb)
Cullin 4A
Death associated transcription factor 1
Caspase 4
Bruton agammaglobulinemia tyr kinase
Cell death-regulatory protein GRIM19
Dudulin 2
Functions of differenciated hepatocyte
Detoxication (down-regulatedb)
Cytochrome P450, IIA, polyp. 6
Cytochrome P450, IVF, polyp. 12
Cytochrome P450, IIIA, polyp. 7
Formyltetrahydrofolate dehydrogenase
Cytochrome P450, IIE, polyp. 1
Cytochrome P450, IIIA, polyp. 4
Metabolism (down-regulatedb)
Glycogen synthase 2 (liver)
Glucokinase regulatory protein
Phosphoenolpyruvate carboxykinase 1
Aldo-keto reductase family 1B1
Plasma protein (up-regulatedb)
Fibrinogen, a polypeptide
Plasma protein (down-regulatedb)
Fibrinogen, b polypeptide
IMAGE
Late FL
HCC
110,503c
309,800
2.59d
2.41
2.49(3)e
2.37(3)
309,748
111,704
366,332
308,551
0.29
0.42
0.26
0.30
0.60(6)
0.57(4)
0.42(5)
0.36(5)
198,542
757,511
112,363
308,473
130,029
83,125
198,622
310,610
0.30
0.22
0.38
0.27
0.44
0.22
0.29
0.31
0.62(4)
0.53(5)
0.49(5)
0.43(5)
0.42(6)
0.41(5)
0.39(5)
0.34(6)
120,306
112,471
198,622
0.38
0.28
0.29
0.54(5)
0.44(5)
0.39(5)
310,431
113,561
126,322
246,748
366,332
321,275
0.48
0.38
0.39
0.41
0.26
0.25
0.64(3)
0.57(4)
0.54(4)
0.53(3)
0.42(5)
0.15(9)
77,451
127,203
121,305
128,680
77,826
83,240
0.39
0.31
0.41
0.34
0.23
0.32
0.65(3)
0.55(4)
0.55(4)
0.47(5)
0.40(6)
0.36(7)
113,358
126,617
742,082
28,882
0.36
0.45
0.32
0.27
0.74(6)
0.62(4)
0.55(5)
0.45(5)
429,555
0.40
2.05(6)
112,334
0.32
0.49(5)
a
The complete data are provided as a supplementary Table S3 on line.
In most instances, a similar regulation was found in late FL and HCC.
c
IMAGE clone number used as a unique identifier for every mRNA.
d
Ratio of the abundance in late FL vs the mean abundance in four control livers.
e
Ratio of the mean abundance in nine HCC samples vs the mean abundance in four control livers (number of HCC samples with a significantly different
abundance vs controls).
b
GRIM-19. Down-regulated mRNAs for proteins of
the translational machinery were for instance: (i)
deoxyhypusine synthase that controls the formation of
the translation initiation factor-5A, (ii) the ribosomal
proteins L13a, L29, S3A, S18 and S26, and (iii) the a1
subunit of translation elongation factor-1 which delivers
aminoacyl tRNAs to ribosomes.
Down-regulated mRNAs for proteins involved in cell
proliferation were, for instance, the DEAD box polypeptide6 believed to be involved in embryogenesis and cell growth
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
and division, peroxiredoxin-1 whose proliferative effect
may be of relevance in cancer, and the IL-6 receptor that
mediates the IL-6-induced cell proliferation. Strikingly, the
mRNAs for apoptosis activators such as cullin 4A,
the death-associated transcription factor 1, caspase 4, the
Bruton’s agammaglobulinemia tyrosine kinase, GRIM19
and dudulin-2 were all down-regulated. Taken together,
these data suggest that limitation of apoptosis predominates
over active proliferation during liver development and
HCC.
mRNAs for proteins that are typical of detoxication,
metabolism and plasma protein production in the differentiated hepatocyte were down-regulated. Detoxication
proteins included various cytochromes P450, and formyltetrahydrofolate dehydrogenase whose down-regulation is
proposed to enhance tumor cell proliferation. Metabolic
enzymes were, for instance, those participating in
glucose/glycogen metabolism, such as glycogen synthase2, the glucokinase-regulatory protein, phosphoenolpyruvate
carboxykinase-1, and aldo–keto reductase 1B1. Among the
plasma proteins, an opposite regulation of the mRNAs for
fibrinogen-a and -b chains was observed in HCC, which is
possibly related to the importance of some fibrinogen
peptides in tumor cell angiogenesis [30]. All other plasma
protein mRNAs were down-regulated.
3.4. Microarray data confirmed by q-RT-PCR
As a control, we selected the six mRNAs with the most
marked and frequent difference in abundance between our
HCCs vs control samples above. The AFP mRNA was also
studied, given the use of blood AFP in the clinical follow-up
of HCC [31]. The relative abundance of these mRNAs was
next determined by q-RT-PCR in a novel set of HCCs and
controls (HCC10–HCC28 and C5–C13 in Table 1) as well
as in our early or late FL. The results shown as a
supplementary Fig. S2 perfectly fit our analysis made by
microarray, including the strong or borderline up-regulation
of the KIAA0789 or AFP mRNA, respectively, as well as
the down-regulation of mRNAs coding for plasma proteins
(haptoglobin, apolipoprotein C3, orosomucoid, albumin) or
other proteins (dudulin-2).
3.5. Dudulin-2 mRNA as a marker of the
cirrhosis-to-HCC transition
As the abundance of dudulin-2 mRNA was strikingly
different in HCCs vs controls, it was further measured in
cirrhotic tissues and found to be down-regulated not only in
the HCC nodules but also in the paired cirrhotic samples.
This feature was found regardless of the tumor differentiation grade (Fig. 4), etiology or vascular invasion (data not
shown). On the contrary, the mean dudulin-2 mRNA level in
livers from HCC-free cirrhotic patients was slightly above
the mean level in controls and no overlap between the levels
in HCC-free vs HCC-associated cirrhotic livers was found.
*
100
$
*
$
$
$
75
mRNA abundance (%)
866
50
ns
p<10–3
p<10–3
25
C
HCC-free CIR T
CIR
ES1
CIR
T
ES2
CIR
T
ES3
CIR T
ES4
Fig. 4. q-RT-PCR of dudulin-2 mRNA. The patients are all those listed
in Table 1. All values (bar: mean) are expressed as a percent of the
highest abundance (100%) found in one of the samples (C, control
liver; CIR, cirrhosis; T, tumor). ES1-4 refers to the tumor differentiation grade 1–4 [46]. The Student’s t test for two samples was used for
comparison of any group vs the control patients (-, P!0.05; $,
P!0.01). The Wilcoxon’s matched-pairs signed ranks test was used for
comparisons of paired samples (significance noted above an horizontal
bracket).
Therefore, repression of dudulin-2 mRNA is associated with
the transition from an HCC-free cirrhosis to a cancerous
liver. Moreover, the relative mRNA levels in hepatocytes vs
non-parenchymal cells pointed to the former as the major
source of dudulin-2 in liver (Fig. S3). Consistent with its
down-regulation in the cancerous hepatocyte, this mRNA
was barely expressed in the tumorous cell lines Hep3B and
HepG2.
4. Discussion
Most transcriptome-oriented studies of HCC in human or
rodents have searched for differences between the cancerous
nodules vs surrounding cirrhotic tissue whereas few
comparisons with control adult liver have been made
[1,3,5,16,17]. Moreover, studies interested in gene downregulation in HCC have focused on a few selected mRNAs
[32]. Therefore, a global tendency to down-regulation of
mRNA abundance in the cancerous hepatocyte has gone unnoticed [3,16,33] or could not be appreciated to its full
extent for a matter of incomplete gene coverage [6,17,34,
35]. Our complete analysis of the liver transcriptome has
now identified for the first time that the cancerous
hepatocyte and the late FL, i.e. after 22–24 weeks of
gestation, exhibit a similar trend towards a preferentially
decreased abundance of numerous mRNAs and share
a functionally relevant subset of them. Genetic polymorphism can influence mRNA level and may skew patient
clustering in transcriptome analysis [8]. This potential
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
drawback was unlikely to plague our conclusions since
hundreds of mRNAs from unlinked genes were found to be
down-regulated in most, if not all, our HCC patients.
Gene mutations or deletions as commonly observed in
the cancerous hepatocyte [37] cannot account for a shared
down-regulation of given mRNAs in HCC and FL, as
genome integrity is retained in the latter. Methylation of
CpG islands which is commonly found during development
and cancer [1,38] and limits the access of transcription
factors to gene promoters is likely to participate in this
shared down-regulation. Moreover, we have now found
that the mRNA levels for several members of the AP-1,
ATF, C/EBP, and XBP families are specifically altered in
HCC and late FL. In keeping with this, AP-1 and XBP-1
mostly act as regulators of the hepatic cell cycle after midgestation [11], the ATF-coactivator CBP is expressed late
during liver development [39] and C/EBPa which is upregulated during the perinatal phase is a hallmark of the
differentiated hepatocyte [9,40]. We have also observed
unusual frequencies of potential binding sites for these
LETFs in the set of down-regulated genes shared in HCC
and late FL. Taken together, our data strongly suggest that
some similar LETF-driven transcriptional controls participate in the shared down-regulation of mRNAs in HCC and
late FL. Elsewhere, we did not observe any shared
regulation of mRNAs for other LETFs such as members
of the HNF-1, HNF-3, HNF-4, HNF-6 or Hex family. This is
most likely explained by an early demand for these LETFs
during liver development [9–11], which makes a significant
change in abundance between late FL vs adult liver
unlikely. All our above observations are fully consistent
with HCC having a similarity with the fetal hepatocyte
mostly after the 22–24th week of gestation.
The 22–24th week of gestation is a critical step as major
metabolic and hematopoietic shifts occur in the liver at this
time [19,20]. Accordingly, we observed that specific
changes in hepatocyte-specific metabolism take place
following this developmental shift. This was also observed
in the cancerous hepatocyte and hence this study extends
earlier and more limited observations [16,33,34],
including some that were mainly focused on metabolism
[17,34] and/or plasma proteins [34,36]. Eventually, mRNA
under-expression in the cancerous hepatocyte results in two
major and final events, namely: (i) a limitation of cell typespecific metabolism and associated secretion of plasma
proteins and (ii) a repression of apoptosis. A reduced rate of
apoptosis in HCC has been suspected by others [34] but a
shared mechanism in FL and HCC is a novel finding. Given
the dynamics of some AP-1-related mRNAs noted in this
study, such a limitation of apoptosis that predominates over
active proliferation in the late stages of liver development
and in HCC is consistent with the possible role of some
AP-1 subunits in the apoptosis/proliferation balance [41].
The down-regulation of dudulin-2 mRNA which codes
for a P53-inducible apoptotic protein [42] also fits a limited
apoptosis. Although this down-regulation may be partly
867
attributable to P53 mutations, its relatively higher incidence
vs the incidence of P53 mutations, particularly in HCC-free
cirrhotic disease, suggests an alternative mechanism.
Dudulin-2 may provide a novel, down-regulated HCC
marker that would complement current up-regulated
markers [3–5,43,44]. As the mechanisms leading to
cirrhosis and HCC are likely to vary with etiology [45],
additional studies should clarify whether our observations
mostly made in patients with an hepatitis C- or alcoholismassociated HCC also hold true in the context of an hepatitis
B-related cancer. Given the current lack of markers for the
cirrhosis-to-HCC transition [45], in the future dudulin-2
may appear to be of interest for the follow-up of HCC-free
cirrhosis.
Acknowledgements
We are indebted to Dr P. Ruminy and F. Parmentier for
help with the Taqman analysis and to G. Caroff for liver cell
separation. C.C. and G.L. are the recipients of a fellowship
from the French Ministry for Research and C.C. and C.D.
are the recipients of a fellowship from Association de
Recherche sur le Cancer and Ligue contre le Cancer
(Section de l’Eure), respectively. This work was supported
in part by grants from Association de Recherche sur le
Cancer, Agence Nationale de Recherche sur le SIDA, and
Institut de Recherches Scientifiques sur les Boissons to
J.P.S. and Ligue contre le Cancer to M.S.
Supplementary data
Supplementary data associated with this article can be
found, in the online version, at doi:10.1016/j.jhep.2005.
01.027
References
[1] Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human
hepatocellular carcinoma. Nat Genet 2002;31:339–346.
[2] Iizuka N, Oka M, Yamada-Okabe H, Mori N, Tamesa T, Okada T,
et al. Differential gene expression in distinct virologic types of
hepatocellular carcinoma: association with liver cirrhosis. Oncogene
2003;22:3007–3014.
[3] Kim JW, Wang XW. Gene expression profiling of preneoplastic liver
disease and liver cancer: a new era for improved early detection and
treatment of these deadly diseases? Carcinogenesis 2003;24:363–369.
[4] Smith MW, Yue ZN, Geiss GK, Sadovnikova NY, Carter VS, Boix L,
et al. Identification of novel tumor markers in hepatitis C virusassociated hepatocellular carcinoma. Cancer Res 2003;63:859–864.
[5] Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting
hepatitis B virus-positive metastatic hepatocellular carcinomas using
gene expression profiling and supervised machine learning. Nat Med
2003;9:416–423.
868
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
[6] Choi JK, Choi JY, Kim DG, Choi DW, Kim BY, Lee KH, et al.
Integrative analysis of multiple gene expression profiles applied to
liver cancer study. FEBS Lett 2004;565:93–100.
[7] Neo SY, Leow CK, Vega VB, Long PM, Islam AFM, Lai PBS, et al.
Identification of discriminators of hepatoma by gene expression
profiling using a minimal dataset approach. Hepatology 2004;39:
944–953.
[8] Llovet JM, Wurmbach E. Gene expression profiles in hepatocellular
carcinoma: not yet there. J Hepatol 2004;41:336–339.
[9] Zaret KS. Regulatory phases of early liver development: paradigms of
organogenesis. Nat Rev Genet 2002;3:499–512.
[10] Costa RH, Kalinichenko VV, Holterman AL, Wang X. Transcription
factors in liver development, differentiation, and regeneration.
Hepatology 2003;38:1331–1347.
[11] Duncan SA. Mechanisms controlling early development of the liver.
Mech Dev 2003;120:19–33.
[12] MacSween RNM, Burt AD, Portmann BC, Ishak KG, Scheuer PJ,
Anthony PP. Pathology of the liver. 4th ed. Orlando: Harcourt Brace;
2001, p. 1–1008.
[13] Hsu HC, Cheng W, Lai PL. Cloning and expression of a
developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer
Res 1997;57:5179–5184.
[14] Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P,
Michalopoulos GK. Beta-catenin antisense studies in embryonic
liver cultures: role in proliferation, apoptosis, and lineage specification. Gastroenterology 2003;124:202–216.
[15] Poon RT, Lau CP, Cheung ST, Yu WC, Fan ST. Quantitative
correlation of serum levels and tumor expression of vascular
endothelial growth factor in patients with hepatocellular carcinoma.
Cancer Res 2003;63:3121–3126.
[16] Tellgren A, Wood TJ, Flores-Morales A, Torndal UB, Eriksson L,
Norstedt G. Differentially expressed transcripts in neoplastic hepatic
nodules and neonatal rat liver studied by cDNA microarray analysis.
Int J Cancer 2003;104:131–138.
[17] Xu XR, Huang J, Xu ZG, Qian BZ, Zhu ZD, Yan Q, et al. Insight into
hepatocellular carcinogenesis at transcriptome level by comparing
gene expression profiles of hepatocellular carcinoma with those of
corresponding noncancerous liver. Proc Natl Acad Sci USA 2001;98:
15089–15094.
[18] Plescia C, Rogler C, Rogler L. Genomic expression analysis
implicates Wnt signaling pathway and extracellular matrix alterations
in hepatic specification and differentiation of murine hepatic stem
cells. Differentiation 2001;68:254–269.
[19] Yu Y, Zhang C, Zhou G, Wu S, Qu X, Wei H, et al. Gene expression
profiling in human fetal liver and identification of tissue- and
developmental-stage-specific genes through compiled expression
profiles and efficient cloning of full-length cDNAs. Genome Res
2001;11:1392–1403.
[20] Kelley-Loughnane N, Sabla GE, Ley-Ebert C, Aronow BJ,
Bezerra JA. Independent and overlapping transcriptional activation
during liver development and regeneration in mice. Hepatology 2002;
35:525–534.
[21] Nagata T, Takahashi Y, Ishii Y, Asai S, Sugahara M, Nishida Y, et al.
Profiling of genes differentially expressed between fetal liver and
postnatal liver using high-density oligonucleotide DNA array. Int
J Mol Med 2003;11:713–721.
[22] Coulouarn C, Lefebvre G, Derambure C, Lequerre T, Scotte M,
Francois A, et al. Altered gene expression in acute, systemic
inflammation detected by complete coverage of the human liver
transcriptome. Hepatology 2004;39:353–364.
[23] Masson S, Daveau M, Francois A, Bodenant C, Hiron M, Teniere P,
et al. Up-regulated expression of HGF in rat liver cells after
experimental endotoxemia: a potential pathway for enhancement of
liver regeneration. Growth Factors 2001;18:237–250.
[24] Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of
microarray data. Bioinformatics 2002;18:207–208.
[25] Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM,
et al. Gene ontology: tool for the unification of biology. Nat Genet
2000;25:25–29.
[26] Zhang H, Ramanathan Y, Soteropoulos P, Recce ML, Tolias PP. EZRetrieve: a web-server for batch retrieval of coordinate-specified
human DNA sequences and underscoring putative transcription
factor-binding sites. Nucleic Acids Res 2002;30:e121.
[27] Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, et al.
TRANSFAC: transcriptional regulation, from patterns to profiles.
Nucleic Acids Res 2003;31:374–378.
[28] Passegue E, Jochum W, Behrens A, Ricci R, Wagner EF. JunB can
substitute for Jun in mouse development and cell proliferation. Nat
Genet 2002;30:158–166.
[29] Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K. C/EBP family
transcription factors are degraded by the proteasome but stabilized by
forming dimer. Oncogene 2003;22:1273–1280.
[30] Staton CA, Brown NJ, Lewis CE. The role of fibrinogen and related
fragments in tumour angiogenesis and metastasis. Expert Opin Biol
Ther 2003;7:1105–1120.
[31] Velazquez RF, Rodriguez M, Navascues CA, Linares A, Perez R,
Sotorrios NG, et al. Prospective analysis of risk factors for
hepatocellular carcinoma in patients with liver cirrhosis. Hepatology
2003;37:520–527.
[32] Kinoshita M, Miyata M. Underexpression of mRNA in human
hepatocellular carcinoma focusing on eight loci. Hepatology 2002;36:
433–438.
[33] Tackels-Horne D, Goodman MD, Williams AJ, Wilson DJ,
Eskandari T, Vogt LM, et al. Identification of differentially
expresssed genes in hepatocellular carcinoma and metastatic liver
tumors by oligonucleotide expression profiling. Cancer 2001;92:
395–405.
[34] Okabe H, Satoh S, Kato T, Kitahara O, Yanagawa R, Yamaoka Y,
et al. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes
involved in viral carcinogenesis and tumor progression. Cancer Res
2001;61:2129–2137.
[35] Delpuech O, Trabut JB, Carnot F, Feuillard J, Brechot C,
Kremsdorf D. Identification, using cDNA macroarray analysis, of
distinct gene expression profiles associated with pathological and
virological features of hepatocellular carcinoma. Oncogene 2002;21:
2926–2937.
[36] Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene
expression patterns in human liver cancers. Mol Biol Cell 2002;13:
1929–1939.
[37] Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer
Biol 2000;10:185–200.
[38] Chiba T, Yokosuka O, Arai M, Tada M, Fukai K, Imazeki F, et al.
Identification of genes up-regulated by histone deacetylase inhibition
with cDNA microarray and exploration of epigenetic alterations on
hepatoma cells. J Hepatol 2004;41:436–445.
[39] Ghoshal S, Pasham S, Odom DP, Furr HC, McGrane MM. Vitamin A
depletion is associated with low phosphoenolpyruvate carboxykinase
mRNA levels during late fetal development and at birth in mice.
J Nutr 2003;133:2131–2136.
[40] Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV,
Wilde MD, et al. Impaired energy homeostasis in C/EBPa knockout
mice. Science 1995;269:1108–1112.
[41] Mikula M, Gotzmann J, Fischer AN, Wolschek MF, Thallinger C,
Schulte-Hermann R, et al. The proto-oncoprotein c-Fos negatively
regulates hepatocellular tumorigenesis. Oncogene 2003;22:
6725–6738.
[42] Passer BJ, Nancy-Portebois V, Amzallag N, Prieur S, Cans C,
Roborel de Climens A, et al. The p53-inducible TSAP6 gene
product regulates apoptosis and the cell cycle and interacts with
Nix and the Myt1 kinase. Proc Natl Acad Sci USA 2003;100:
2284–2289.
C. Coulouarn et al. / Journal of Hepatology 42 (2005) 860–869
[43] Iizuka N, Oka M, Yamada-Okabe H, Mori N, Tamesa T, Okada T,
et al. Comparison of gene expression profiles between hepatitis B
virus- and hepatitis C virus-infected hepatocellular carcinoma by
oligonucleotide microarray data on the basis of a supervised
learning method. Cancer Res 2002;62:3939–3944.
[44] Hippo Y, Watanabe K, Watanabe A, Midorikawa Y, Yamamoto S,
Ihara S, et al. Identification of soluble NH2-terminal fragment of
869
glypican-3 as a serological marker for early-stage hepatocellular
carcinoma. Cancer Res 2004;64:2418–2423.
[45] Bruix J, Boix L, Sala M, Llovet J. Focus on hepatocellular carcinoma.
Cancer Cell 2004;5:215–219.
[46] Edmondson HA, Steiner PE. Primary carcinoma of the liver: a
study of 100 cases among 48,900 necropsies. Cancer 1954;7:
462–503.