Experiment 3. The Separation of Solids, Recrystallization and

Experiment 3. The Separation of Solids, Recrystallization and Melting Point Determination.
References: Eaton, Laboratory Investigations in Organic Chemistry
Pasto and Johnson, Organic Structure Determination , pp. 57-65.
Adamson, Textbook of Physical Chemistry , pp. 395-396.
CRC Press, Handbook of Tables for Organic Compound Identification
INTRODUCTION:
The separation of a mixture of two or more substances is a problem which is frequently encountered in organic chemistry. Experiment 2 demonstrated how to separate and isolate organic liquids, while Experiment 3 deals with the separation/isolation of solids by liquid-liquid extraction. In this experiment, you are provided with a mixture of 4-chloroaniline, and benzoic acid in approximately equal proportions, along with some impurities. After separating 4chloroaniline and benzoic acid from the mixture, you will purify one of the components by crystallization, and identify both of them by their melting points and IR spectra.
NH
2 O OH
Cl
4-Chloroaniline Benzoic Acid
In this lab, you will learn a few techniques that will apply to many of the upcoming experiments.
BACKGROUND:
Technique A: Separation of Solids by Liquid-Liquid Extraction
The separation of a mixture of chemical compounds A and B into its separate constituents is achieved by methods which depend on a difference in either the physical or chemical properties of A and B. If A and B differ in their physical properties then they may usually be separated by virtue of their different solubilities in a pair of immiscible solvents. An example of such a mixture would be sodium chloride and cholesterol, both of which are white, crystalline solids.
The separation here would be quite simple. Sodium chloride, an ionic salt, is soluble in water but insoluble in ether whereas cholesterol, a neutral organic molecule, is soluble in ether and only very sparingly soluble in water. Shaking the mixture with a mixture of water and ether would result in the formation of two immiscible solutions, one of sodium chloride in water and one of cholesterol in ether. Separation of the two solutions in a separatory funnel and the evaporation of each solution separately would then lead to the recovery of individual samples of both sodium chloride and cholesterol.
In a more complex case, such as the separation of sodium chloride from 3-aminobenzoic acid where one component (in this case the 3-aminobenzoic acid) is soluble in both water and ether, a process known as extraction has to be used. There the mixture is dissolved in water and the resulting solution shaken with ether in a separatory funnel. The 3-aminobenzoic acid, being soluble in both the aqueous and ethereal layers, will partition itself between the two layers and this partitioning is described by the partition or distribution coefficient, K p
, as follows:
K p
= solubility in ether / solubility in water
The solubility of 3-aminobenzoic acid in water at 20 o
C is 6.0 gram/litre (g/L), while in ether it is
20.0 g/L. Therefore,
K p
= (20 g/L) / (6 g/L) = 3.33
Suppose that 2.0 g of 3-aminobenzoic acid is present in the mixture and that the compound is allowed to partition itself between l litre of water and l litre of ether at 20
o
C. How much of the acid will be present in each layer?
If Z is allowed to represent the concentration in g/mL of acid in the water, then from the definition of the partition coefficient,
K p
= (concentration of acid in ether) / Z = 3.33,
Thus, (concentration of acid in ether) = 3.33 x Z wt. of acid in 1 L of water = l000 x Z wt. of acid in 1 L of ether = l000 x 3.33 x Z
But, total wt. of acid = 2.0 g
Therefore,
(l000 x Z) + (3.33 x l000 x Z) = 2.0 g i.e. 4330 x Z = 2.0 g or Z = 2.0 / 4330 = 0.000462 g/L
Therefore, wt. of acid in water = 1000 x Z = 0.462 g and wt. of acid in ether = 1000 x 3.33 x Z = 1.538 g
Thus, by a single extraction of 1000 mL of aqueous solution by 1000 mL of ether, 1.538 g of the original 2 g of acid present in the water will be transferred to the ether layer. It should now be evident that by removal of the first litre of ether and re-extraction of the aqueous layer with a fresh batch of ether, partitioning of the remaining 0.462 g of acid between the aqueous and the second ethereal layers will result in the transfer of a further 0.355 g of acid into the ether, leaving only 0.107 g in the aqueous layer. The repeated extraction of the aqueous layer with fresh batches of ether will lead therefore to the removal of most of the acid into the ether. The acid may then be recovered by evaporation of the combined ether extracts.
In many cases, mixtures of organic compounds can be separated by methods which depend on a difference in their chemical properties. Frequently, one or more compounds of a mixture is transformed into a salt for easy separation. Salts, in general, are water soluble and insoluble in organic solvents.
For example, for acidic compounds (HA):
HA + Na
+
OH
-
----------> Na
+
A
-
+ H
2
O
For bases (B:):
B: + H
3
O
+
----------> BH
+
+ H
2
O
In this experiment, the mixture may be readily separated by virtue of the fact that 4-chloroaniline will react with HCl in water and benzoic acid will react with sodium bicarbonate (NaHCO
3
) in water. Thus, if a solution of a mixture of 4-chloroaniline, and benzoic acid in ether is shaken with aqueous HCl the amine will be removed into the aqueous layer as its ammonium salt, whereas benzoic acid and the impurities will remain dissolved in the ether.
With only benzoic acid and impurities left in the ether, the benzoic acid then can be extracted as its sodium salt by extracting the ether layer with aqueous NaHCO
3
.
O OH O O
a
+ aHCO
3
+
+ H
2
O + CO
2
After separation of the aqueous and etheral layers, the 4-chloroaniline can be regenerated by basification with aqueous NaOH, while benzoic acid can be regenerated from its sodium salt by acidification with dilute acid.
When performing these extractions, you will have two heterogeneous layers; an organic and an aqueous layer. As a general rule, the organic layer will be the top layer, except for organic
2
Cl
2
, CH
3
CCl
3
) will be on the bottom layer, since they are denser that water.
Products from an organic reaction are seldom obtained in a pure state directly from the reaction rystallization from a suitable solvent.
As was previously pointed out, different compounds have varying solubility in different solvents.
We will use this principle coupled with the effect temperature has on solubility in the technique
. The process of crystallization depends on the fact that most organic compounds are more soluble in hot solvents than in cold, and that the impurities present will
1) Dissolving the solid mixture in a minimal volume of a solvent, usually at the boiling point of the solvent. [NOTE: Insoluble impurities are removed by filtering the hot solution at its boiling point.] osit crystals of the compound. Crystallization may need by "seeding" the cold
3) Filtering the crystals from the liquid solution (called th mother liquor ). While filtering, the by recystallization in water?
Using 100 mL of water, the solid could all dissolve at 100°C. A minimum amount of solvent is used in recrystallization procedures so that the greatest amount of solid can be recovered .
Upon cooling the solution to 25°C, benzoic acid crystallizes in an amount equal to the quantity that was dissolved at 100°C (5.7 g) minus the quantity that is still soluble in 100 mL of water at
25°C (0.3 g):
Recrystallized Benzoic Acid = 5.7g - 0.3g = 5.4g
NOTE: Some benzoic acid is still soluble at 25°C, which is why we use a minimal amount of solvent.
Likewise, some phthalic acid will still be soluble at 25°C, therefore all the phthalic acid (0.3 g) will remain in the water solution. The sodium chloride will also be completely dissolved in the water solution. If there is any dibromobenzene impurity, then practically no dibromobenzene will dissolve at any temperature and so it should have been removed from the water solution when all the other solids are dissolved (at the boiling point of the water). After the dibromobenzene has been removed and the solution has cooled, we can now filter out the pure benzoic acid.
Properties of an Ideal Recrystallizing Solvent
If recrystallization is to be effective, the solvent must be properly selected. A good recrystallization solvent should:
(a) dissolve a moderate quantity of the substance to be purified at an elevated temperature, but only a small quantity at low temperatures
(b) not react with the substances to be purified
(c) dissolve impurities readily at a low temperature or not dissolve them at all
(d) be readily removable from the purified product. This usually means that the solvent must have a relatively low boiling point and evaporate readily.
How to Find a Good Recrystallizing Solvent
Let us assume that you have an unknown compound and you want to purify it by crystallization.
First, determine its solubility properties in simple available solvents.
It is usual to try water (seldom successful), methanol, ethanol, acetic acid, petroleum ether
(available in several boiling ranges) and, less commonly, ether, chloroform, ethyl acetate, acetonitrile and dimethylformamide. Add about 20 mg of material to 1 ml of cold solvent. Shake up to 3 min to help dissolution. If insoluble, warm the solvent to boiling. Note the solubility properties in each solvent (cold and hot). Ideally your material should be insoluble in cold solvent and soluble in hot solvent; if you find a solvent with these properties you can stop here as this will probably be a suitable solvent for recrystallization.
If no single solvent is found suitable, then a mixed solvent recrystallization is in order. For this you require two miscible solvents . For mixed solvent recrystallization your material should be relatively soluble in one solvent and relatively insoluble in another solvent.
For example, a substance which is very soluble in alcohol and almost insoluble in water may crystallize well from a mixture. The correct procedure is to dissolve the solid in the minimum amount of boiling alcohol and add warm water drop wise . Each drop will produce a cloudiness which at first clears on mixing. When the warmed solution just fails to clear on shaking, a few drops of alcohol are added, the mixture is re-heated and set aside for crystallization. Any insoluble impurities are best removed by filtering the hot alcohol solution before adding water.
Technique C: Melting Point Determination
A melting point can be used to identify a substance and to get an indication of its purity. The melting point (or freezing point) of a solid is the temperature at which the solid exists in equilibrium with its liquid state under an external pressure of one atmosphere. More precisely, it is the temperature at which the vapor pressure of the solid phase becomes equal to the vapor pressure of the liquid phase . Experimentally, it is extremely difficult to establish the exact temperature at which this equilibrium is established; therefore, the temperature range over which liquid and solid are found to coexist is called the melting point. For example, a solid may be reported to have a `melting point' of 100-101 droplet of liquid was observed at 100 o o
C; this means that, on heating slowly, the first
C and the last crystal of solid disappeared at 101 o
C.
Both the melting point range (the interval between the beginning of liquefaction and complete liquefaction) and the temperature of complete liquefaction are valuable indicators of the purity of the solid compound . A pure crystalline organic compound usually possesses a sharp melting point and it melts completely over a narrow temperature range of not more than 0.5-1.0
o
C, provided good technique is followed. The presence of even small amounts of impurities usually produces a depression of the temperature at which melting is complete and usually produces a marked increase in the width of the melting point range . A wide melting point range usually indicates that a substance is impure, but it may also result from the fact that the pure substance undergoes some decomposition prior to reaching its melting point. In some cases, the material undergoes a slight liquefaction and contraction at a temperature below the true melting point; in others, the material may decompose and discolour so badly that a definite melting point cannot be observed .
Theory of Melting Point Depression
The theory underlying the solid/liquid change of state is based primarily on the phenomenon of vapor pressure. Both liquid and solid forms of a compound exert vapor pressure; vapor pressure increases with increasing temperature but that of the solid increases more rapidly than that of the liquid . At the melting point, the vapour pressures of the solid and liquid phases are equal. A soluble impurity contributes to the total vapour pressure, therefore lowering the partial vapour pressure required of the pure substance in the melt and thus lowers the temperature necessary for melting. [Recall (from Exp. 2): Dalton's law states that the total vapour pressure of a liquid solution is the sum of the partial pressures of the components].
Additions of more and more impurity will produce corresponding lowering in the partial vapor pressure of the pure substance and hence, lowering of the melting point. Finally, however, a limiting point is reached at which the impurity concentration is just sufficient to `saturate' the liquid; any additional impurity does not dissolve and cannot further depress the melting point.
This point is known as the Eutectic Point and the limiting temperature is called the Eutectic
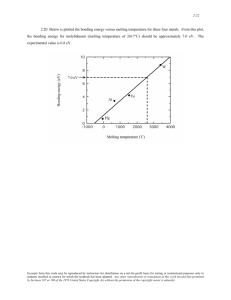
Temperature and the composition of the melt, the Eutectic Composition. Alternatively, the eutectic temperature can be described as the temperature below which a mixture of C and D cannot exist as a liquid; or, the temperature at which C and D can co-crystallize from the liquid melt (see Figure 2).
Eutectic Point
The nature of the eutectic point and, more importantly, its influence on the observed melting point range are more effectively illustrated by a generalized, equilibrium temperature versus composition diagram, Figure 2. For any concentration, the curve (points C to E to D in Figure 2)
e temperature at which the last traces of solid will melt, when heated (or the temperature at which the first traces of solid will crystallize if the liquid mixture is cooled).
Point Composition Diagram for a Mixture of C + D
As the solid is heated and the temperature passes the eutectic melting point temperature (the line at temperature, T
E
), the solid starts to melt. The initial liquid melt will have the same solid melts at the eutectic composition, until all of the impurity has entirely melted. Upon further heating, the remaining pure solid will begin to melt.
For example, using Figure 2, consider a mixture of 80% C and 20% D. When heat is applied, the ature of the solid mixture will rise; no change in the physical state will occur until the eutectic temperature (T
E E
, the solid will melt to form a liquid phase of eutectic ntirely melted. At this point, the remaining C will melt with the last crystal melting at the temperature, T
A
(below the melting point of pure C). Even though the remaining solid is pure C, the vapour pressure of the solid um with the vapour pressure of the impure liquid (impurities
Therefore, in theory , then, for any solid compound containing a relatively small amount of impurity, gin at the eutectic temperature and be complete at some temperature lower than the melting point of the pure compound . Moreover, if the concentration of the impurity were increased, the upper limit of the melting would be lowered and therefore the melting range decreased.
However, in practice it is extremely difficult to detect the initial melting or eutectic conditions,
, a nearly pure solid compound tends to show a narrow melting range with an upper
limit near the true melting point whereas a rather impure compound usually shows a broader melting range with the maximum temperature considerably below the true melting point.
It should be noted that a sample whose composition is exactly that of the eutectic mixture will exhibit a sharp melting point, melting completely at the eutectic temperature. Thus a eutectic mixture may sometimes be mistaken for a pure compound. One should also note that the temperature versus composition diagram may not look like Figure 2, since some mixtures of compounds have more than 1 eutectic point and even a few mixtures have no eutectic point.
PRE-LAB PREPARATION:
Read the experimental procedure so that you are prepared for the lab and you understand the safety and disposal information for the chemicals you are using in this experiment. Wear gloves and change them often.
1 . If 3.0 g of a solid is dissolved in 300 mL of water and is to be extracted by 300 mL of chloroform (K p
= 8), calculate the weight of solid present in each layer.
2 . What effects do impurities have on the melting point of an organic compound?
3 . Draw a flow chart (see the top of the next page for the start of a typical flow chart) that represents the entire extraction procedure in this experiment as well as the final steps of recovering the products involving NaOH and HCl.
EXPERIMENTAL PROCEDURE:
You should be careful in this Experiment to obtain as quantitative a separation of the mixture as possible. DO INDIVIDUALLY
Safety and Disposal Data for Compounds to be Distilled and Test Reagents.
Compound
Benzoic Acid
Mol. Wt. (g/mol) Safety and Disposal Data
122.12 Harmful if swallowed. Irritating to eyes.
Dispose in ‘Solid Waste’ Container.
4-chloroaniline
Diethyl Ether
2M & 6M (HCl)
Hydrochloric Acid
5% Sodium
Bicaronate
10% Sodium
Hydroxide (NaOH)
127.57
74.12
36.46
84.00
39.99
Toxic by inhalation, in contact with skin and if swallowed. Dispose in ‘Solid Waste’ Container.
Irritant. Extremely flammable. May form explosive peroxides. Dispose in Organic Waste.
Irritating to eyes, respiratory system and skin.
Use gloves , especially when handling 6M HCl.
Dispose 6M HCl in ‘Inorganic Acids and Salts’.
Avoid contact with skin and eyes.
Corrosive. Causes burns. Wear gloves when handling. Dispose in ‘Inorganic Bases’.
Equipment Used
All the equipment needed to perform this experiment should be in one of the three drawers in the top row of your workstation, one of the drawers underneath your fumehood or the common counter for your TAs group.
Only cap when shaking, leave open all other times!!
Ether (Same throughout all extractions) you may need to add more ether to the layer as it evaporates easily
Part A : HCl /Water
Part B : Sodium Bicarborante (NaHCO
3
)/Water
Separatory Funnel Setup for this lab
Part A: Separation of 4-Chloroaniline from the Mixture
Support Ring ( Clamp to metal scaffolding in fumehood ) To hold separatory funnel
1. Liquid-Liquid Extraction with an Acid a) Weigh out 1.0 g of the mixture of benzoic acid and 4-chloroaniline and transfer the solid to a
125 mL Erlenmeyer flask. Add to the flask 10 - 15 mL of ether and swirl until the solid is dissolved. Transfer the solution to a 60 mL separatory funnel, washing out the Erlenmeyer flask with a small volume (ca. 3-5 mL) of ether (use a Pasteur pipette) to ensure that no solid remains in the flask. b) Then add carefully to the separatory funnel 15 mL of 2 M HCl ( NOT 6M!!!). Stopper the funnel and shake it carefully, remembering to release the pressure caused by the heat of the reaction at frequent intervals through the tap. When shaking is complete, remove the stopper and set the funnel on a clamp or a support ring, to allow the two layers to separate. You should
NEVER leave a separatory funnel sealed , gas builds up, and it becomes pressurized!!!
[NOTE: Ether has a boiling point (bp) of only 35 o
C; some of your ether may evaporate during the extraction process. If this occurs (as you will see by your upper layer being very small in volume) simply add 5 -10 mL of more ether.] c) When separation is complete, remove the stopper and carefully run off the lower aqueous layer into a clean 125 mL Erlenmeyer flask, and then add a further 15 mL (approx.) of 2 M HCl to the remaining ether solution in the separatory funnel and repeat the extraction procedure. The second 15 mL of aqueous solution may then be run off and combined with 15 mL obtained from the first extraction. d) The ether layer should now be washed by adding to it 15 mL of water, shaking as before and then running off the aqueous washings into the combined aqueous (HCl) extracts. The resulting aqueous solution ( total of approx. 45 mL ) should then be labeled "4-chloroaniline, hydrochloride salt" and used in the next step.
2. Recovery of 4-Chloroaniline a) The solution of 4-chloroaniline hydrochloride must now be treated with base to recover the free amine (as a solid). Add, a bit at a time and with stirring, 10% NaOH solution to your
Erlenmeyer containing the 4-chloroaniline hydrochloride (approx. 30-35 mL of the 10% NaOH); cool your flask in cold water if necessary. Check the pH with pH-paper in the latter stages of the addition to ensure that the pH of the solution becomes 11-12. b) Before filtering the precipitated 4-chloroaniline, you should decant the solution (See photo). This will allow you to remove the majority of the liquid so you filter less. To do this, let the beaker sit until most of the crystals are sitting immobile at the bottom of the beaker. Then carefully place a glass rod across the beaker and slowly pour the liquid into another beaker without allowing the solid to pour out. Once you have removed about ¾ of the original starting liquid, you can collect the crystals by vacuum filtration on a Hirsch funnel. Refer to exp. 2 to remind yourself how to do vacuum filtrations. c) The precipitate is then washed with a little cold water, dried on a sheet of filter paper and weighed. d) Describe the crystals (colour, shape). Calculate the % wt of 4chloroaniline present in the mixture. Save your sample for PART C where you will determine its melting point.
NOTE : While the solution is filtering, you can get started on the separation of benzoic acid
(Part B).
Part B: Separation of Benzoic Acid from the Mixture
1. Liquid-Liquid Extraction with a Base a) Now add carefully to the separatory funnel ( still containing your ether layer ) 15 mL of 5% sodium bicarbonate solution (NaHCO
3
). Stopper the funnel and shake it carefully, remembering to release the pressure caused by carbon dioxide evolution at frequent intervals through the tap. When shaking is complete, remove the stopper and set the funnel on a clamp or a support ring, to allow the two layers to separate. You should NEVER leave a separatory funnel sealed , gas builds up, and it becomes pressurized!!! b) When separation is complete, run off the lower aqueous layer into a second clean 125 mL
Erlenmeyer flask, and then add a further 15 mL of 5% sodium bicarbonate solution to the remaining ether solution in the separatory funnel and repeat the extraction procedure. The second
15 mL of aqueous solution may then be run off and combined with 15 mL obtained from the first extraction. c) The ether layer should now be washed by adding to it 15 mL of water, shaking as before and then running off the aqueous washings into the combined aqueous extracts. The resulting aqueous solution (total of approx. 45 mL) should then be labelled "benzoic acid, sodium salt" and used in the next step.
2. Recovery of Benzoic Acid a) The solution of benzoic acid as the sodium salt must now be treated with dilute acid to recover the benzoic acid. To do this, simply add dilute ( 6M ) hydrochloric acid (NOT 2M)
SLOWLY and carefully to the solution, with constant swirling, until a dense, white precipitate of benzoic acid is apparent and the solution is acidic to Congo Red indicator paper (it turns blue in acid at about pH 2). During this addition, carbon dioxide is evolved and care is necessary in order to prevent the solution from frothing out of the flask .
b) Before filtering the precipitated benzoic acid, you should decant the solution (See recovery of
4-chloroaniline). Once you have removed about ¾ of the original starting liquid, you can collect the crystals by vacuum filtration on a Hirsch funnel, wash them with a small amount of cold water and dry them on a sheet of filter paper before weighing. c) Describe the crystals (colour, shape). Calculate the % of benzoic acid present in the mixture.
[If you obtained little, or no, precipitate of benzoic acid you probably did not extract the mixture with NaHCO
3
correctly and had too dilute a solution - so repeat the extraction before moving on.]
3. Recrystallization of Benzoic Acid with Water
The benzoic acid may not be very pure and so it will need to be recrystallized. a) SET ASIDE a small amount of CRUDE material for determination of its melting point.
b) To a 125 mL Erlenmeyer flask containing the rest of the isolated benzoic acid, add about 10 mL of hot water and place the flask on a hot plate. DO NOT ADD THE BENZOIC ACID TO
THE WATER!! c) Heat the solution to a gentle boil and if the solid does not dissolve completely add hot water in
SMALL portions until all the benzoic acid dissolves. After each addition of water bring the solution back to the boiling point; this process takes a lot less time if you already have water at or near the boil in another flask on the hot plate. Continue until all the benzoic acid has dissolved.
d) When all the benzoic acid has dissolved and the solution is clear, set the flask aside to cool to room temperature slowly for crystallization. After crystallization is complete at room temperature, cool your flask in an ice-bath for a few minutes (do this particularly if you have very few benzoic acid crystals) e) Before filtering you should decant the solution (as in part 2) then collect the crystals by vacuum filtration on a Hirsch funnel, wash them with a small amount of cold water and dry them on a sheet of filter paper before weighing. f) Describe the crystals (colour, shape). Calculate the % recrystallized of your benzoic acid
(final/initial x 100%). Save your sample for PART C where you will determine its melting point.
Part C: Characterization of 4-Chloroaniline and Benzoic Acid
1. Melting Point Determination – Do in Partners MELTING POINT ONLY
You will perform 3 melting point determinations; 4-chloroaniline, crude and recrystallized benzoic acid. (Run all three at the same time in ONE melting point apparatus) a) Fill a melting point tube with the sample of interest by thrusting the open end into the powder several times and vigorously tapping the sealed end on the table or lightly draw a file across the tube held loosely in the hand. Repeat the procedure until the tube contains a 3 mm column of densely packed powder in the bottom. The melting point range is influenced not only by the purity of the material but also by the size of the crystals, the amount of material, the density of its packing in the tube, and the rate of heating the hot stage. b) Place the capillary in the melting point apparatus, turn on the power and allow the hot-stage temperature to rise. Observe and record the melting range.
Remember : During the determination of the actual melting point range, the temperature should not rise more rapidly than 2 or 3 o
per minute. Heating too quickly will result in an observed range higher than the true one. c) Two temperatures are recorded, the temperature at which the substance begins to liquefy and that at which it becomes completely liquefied. The observed melting point range is the interval between these two temperatures. The behaviour of a material on melting should be observed and recorded carefully, as, for example: melts sharply at 89.0-89.5
o
C; or mp 131-133 o
C, with decomposition; or discolours at 65 o
C, melts slowly at 67-69 o
C.
You should expect melting point ranges for the two compounds to be somewhere between 55-
140 o
C.
TURN OFF MELTING POINT APPARATUS when you are done!
Place the remainder of your samples in the “Solid Waste” bottle provided.