Word file (3225 KB )
advertisement
")
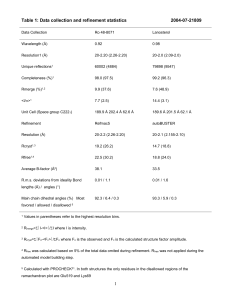
Supplementary information to Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin Hans Jürgen Sass*, Georg Büldt*, Ralf Gessenich*, Dominic Hehn*, Dirk Neff*, Ramona Schlesinger*, Joel Berendzen† & Pal Ormos‡ * Institute of Structural Biology, Research Centre Jülich, 52425 Jülich, Germany † Biophysics Group, Mail Stop D454, Los Alamos Laboratory, Los Alamos, NM 87545, USA ‡ Institute of Biophysics, Biological Research Centre of the Hungarian Academy of Sciences, Szeged, P.O.Box 521, 6701 Szeged, Hungary Additional Results To illustrate the effect of changes in the tertiary structure together with the reorientation of side chains in our M2 state structure the figure below displays the cavities within the molecule of the ground state and the M intermediate. The cavities were determined with the program 1 GRASP using a probe radius of 1 Å. The most important changes are found in the cytoplasmic part of the molecule (top). In the M2 state model a cavity system, connected to the protein surface by a tube (too narrow to allow the passage of water molecules in a rigid bR molecule) has established which could allow a fluctuating water molecule to access the large cavity close to the amino acid Asp96, as described in the main text. Figure : The images of the BR model (a) and the M2 model (b) display the cavities in yellow and the indentation from the outside surfaces in blue, as determined with a probe radius of 1 Å. The ground state (a) has a number of cavities but no clear connections to the cytoplasmic or extracellular surfaces. In the M2 state structure (b) extended cavities are detected in the cytoplasmic domain, necessary for proton transfer from Asp96 to the Schiff base nitrogen of the retinal and for the reprotonation of Asp96 from the cytoplasmic surface. 2 Table 1 Diffraction data Data set Space group Cell dimensions (Å) Resolution (Å) Unique reflections, all Unique reflections, I > 2 Completeness (%), all Completeness (%), I > 2 Rsym (%) ¶ Mosaicity (°) Twinning (%) Illuminated P63 a=b=61.08, c=110.40 20 - 2.25 10792 9284 97.2 20.0 - 2.25 Å 99.4 2.33 - 2.25 Å 83.6 20.0 - 2.25 Å 62.4 2.33 - 2.25 Å 8.2 20 - 2.25 Å 32.8 2.33 -2.25 Å 0.6 38 ground state P63 a=b=60.81, c=110.64 20 - 2.0 14913 13992 94.9 20.0 - 2.0 Å 89.1 2.07 - 2.0 Å 89.0 20.0 - 2.0 Å 71.0 2.07 - 2.0 Å 4.7 20.0 - 2.0 Å 31.9 2.07 - 2.0 Å 0.79 44 ¶ Rsym(l) = ΣhklΣiIhkl,i - Ihkl/ΣhklΣiIhkl,i, with the average intensity lhkl of the multiple observations lhkl,i for symmetri-related reflections. Table 2 Refinement statistics Data set M occupancy (%) § Test set (%) # R factor (%) || Rfree (%) & Illuminated 35 5.3 16.7 20 - 2.25 Å 23.6 20 - 2.25 Å ground state 0 5.5 17.9 20.0 - 2.0 Å 20.8 20.0 - 2.0 Å r.m.s. bond distance dev. (Å) $ r.m.s. angle dev. (°) $ 0.008 1.19 0.006 0.98 Average B factor (Å2) 24.8 / 20.9 31.4 § The BR model with an occupancy of 65% was fixed during refinement. # structure factors for Rfree calculation selected in several thin resolution shells to avoid bias due to the presence of merohedral twinning. || R factor = ΣhklFo - Fc/ ΣhklFo, Fo and Fc are observed and calculated structure factors, respectively. & Rfree = ΣhklFo - Fc/ ΣhklFo, R factor of the test set omitted from the refinement $ root-mean-square deviation. two B factors are given for the illuminated crystal : M model / ground state model. Deposited in the Protein Data Bank: 1CWQ Methods This is a more complete version than in the main text where space limitations prevented the full description. Crystallisation and data collection Crystals were grown according to the protocol of Landau and Rosenbusch 2. Diffraction experiments at 95 K were performed at the X8C beam line of the NSLS Brookhaven with a wavelength = 1.214 Å. Illumination of the crystals bR ground state: The crystal was flash-cooled in the light-adapted ground state to liquid nitrogen temperature. Small amounts of the K intermediate, possibly produced by the room light, were driven back to the BR ground state by illumination with red light of λ = 685 mm. Formation of M intermediate: To generate the late intermediates of the photocycle it is critical to consider that the protein conformation, shown to be correlated to effective proton pumping 3, can be observed only above 260 K . Our procedure for M state formation was: (i) Heating the crystal to room temperature by blocking the nitrogen cold stream. (ii) Illuminating with green light (33 mW, 514 nm) for 1 second. (iii) Cooling by unblocking the cold stream while the green light is still on. (iv) 1 second after cooling has started, the illumination is turned off. (v) Thereafter illuminating with weak red light, to drive the molecules possibly trapped in the K intermediate back to BR. Microspectroscopy of the crystals: Spectroscopic control experiments under identical conditions were performed with a microscope interfaced to a spectrometer. Difference spectra in the visible region were produced by measuring single beam spectra before and after illumination. The amount of components in the photoproduct was estimated on the basis of known spectra of the intermediates 4. On average 60 to 70 % bR molecules in each crystal were accumulated in the M state, small amounts in the N intermediate and 30 to 40 % in BR state. For the ground state it was verified that more than 90 % of bR were driven to the light adapted ground BR state. Treatment of Diffraction Data Determination of lattice parameters (P63 : = = 90°, = 120°; ground sate aBR = bBR = 60.81 Å, cBR = 110.64 Å; illuminated aI = bI = 61.08 Å, cI = 110.40 Å), integration, scaling and merging of the intensities (ground state 20-2.0 Å, Rsym = 4.7 %, illuminated 20-2.25 Å, Rsym = 8.2 %) were done using the HKL suite 5 (Table 1). Conversion to amplitudes was performed with the programs TRUNCATE and MTZUTIL of the CCP4 program package 6. CNS 7, version 0.4, was used for most of the further processing of the data. For quality evaluation about 5 % of the data were selected with the program xdlDATAMAN 8 in resolution shells to be used only for calculation of the Rfree-value. The twinning ratio was first determined by using the twinning server at UCLA university (http://www.doe- mbi.ucla.edu/Services/Twinning/, Yeates, T.O.) and then refined by inspecting the quality of the Rfree after rigid body refinements with different twinning ratios. The used values of the twinning ratio are 0.44 for the ground state crystal and 0.38 for the illuminated one. To cope with the problem of twinning, the available CNS version was slightly modified in the way that for any comparison of measured amplitudes with model amplitudes a twinned version of the model amplitudes is used and for difference calculations, in addition, the intensities were detwinned with the known equations 9 before used in density calculations. Ground state refinement: As a starting model the one of Luecke et. al. 10, PDB:1BRX, was used in normal molecular replacement and rigid body refinement calculations with CNS. The model of the ground state was then further refined through several circles of simulated annealing calculations (using 4000 K and the standard slow cool protocol 11 ) and model building with the molecular graphics program 12 O to complete the E-F loop, to extend the Cterminus to amino acid residue Ser239 and to reduce the model bias. In the beginning retinal and water molecules were not included in the refinement. Residues Arg82, Asp85, Asp96, Glu194, Glu204 and Lys216 were omitted and their conformations carefully checked. At a later stage of the refinement, water molecules were selected with the standard input in CNS and especially the internal ones were checked during further refinement for proper density. Elongated densities in the lipid region were assigned to carbon chains of various length to simulate possible lipid or detergent molecules but without trying to determine real models of lipids or detergents. The final R-values are R = 17.9 % and Rfree = 20.8 %, corresponding to an estimated coordinate error using Rfree of r = 0.211 Å, from the Luzzati plot, and r = 0.131Å, from the SIGMAA analysis, as determined with standard input-files of CNS. 5 M state refinement: The BR model was first oriented in the new unit cell with a molecular replacement calculation and a rigid body refinement. Six different approaches have been used for cross-checking the molecular alterations found for the M2 intermediate: (i) Fourier difference density maps (FoI - FoBR , of the observed structure factors of the illuminated and the ground state crystal) at different resolutions have been calculated and used to locate the changes in the molecule visually with the graphics program 12 O. (ii) A model of two complete bR molecules including water molecules and hydrocarbon chains, one declared as M and one as BR was refined against the data. During the energy minimization refinement the atomic positions of the BR model were fixed whereas the atomic coordinates of the M model with the complementary occupancy were allowed to change. In the same way simulated annealing calculations with standard slow cool protocol 11 at 2500 K and 4000 K were carried out which resulted in M state models comparable to that obtained from simple energy minimisation. Larger changes were observed in the loop regions. (iii) The observed structure factors FoI of the illuminated crystal contain a fraction s of the BR state and a fraction 1-s of the M state. Therefore, extrapolateded difference density maps 13 Δρex = (│FoI│ - s│FcBR│)exp(φcBR) ∕ (1-s) were calculated using amplitudes FcBR and phases φcBR of the ground state model. These maps should show the electron density of the M intermediate for the correct fraction s of the ground state contribution to FoI. (iv) Omit density maps 14 Δρexo were calculated by the Δρex formula using phases φcBR determined without including the retinal and/or Arg82, Asp85, Glu194, Glu204, Lys216, Phe219 and all water molecules (Fig. 2a). These maps were used for visual comparison of the M state conformations of these residues with the resulting densities. (v) Complete omit density maps Δρo were calculated with φcBR as well as FcBR determined from a BR model without the above mentioned amino acids and/or the retinal. Varying fractions of the M and BR state conformations of the omitted residues were correlated to the Δρo densities to define the fraction of the M conformation. The correlations resulted in about 65 % 13-cis retinal and only about 35 % M state conformation for the respective amino acid side chains. If we consider that the M intermediate consists of the early and the late M state contributions and assume that the early M state structure of these residues are very similar to their BR state conformations as verified at low resolution, one would draw the conclusion that on average 35 % of bR molecules in an illuminated crystal are in the ground state, another 35 % in the late M state and 30 % in the early M. (vi) Selecteded difference density maps Δρs = (│FoI│ - 0.65│FcBR│)exp(φcM) ∕ (1-0.65) were calculated using amplitudes FcBR of the ground state model and phases φcM of the M intermediate model (Fig.2b). These maps show 6 the final electron density of the M intermediate. The final R-values of the M state model, including the fixed ground state model, are R = 16.7 % and Rfree = 23.6 %, corresponding to an estimated coordinate error using Rfree of r = 0.279 Å, from the Luzzati plot, and r = 0.239 Å, from the SIGMAA analysis, as determined with standard input-files of CNS. The refinement statistics is summerized in Table 2 and in the header of the Protein Data Bank file 1CWQ. Reference List 1. Nicholls, A., Sharp, K.A. & Honig, B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11, 281-296 (1991). 2. Landau, E.M. & Rosenbusch, J.P. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci U S A 93, 14532-14535 (1996). 3. Ormos, P. Infrared spectroscopic demonstration of a conformational change in bacteriorhodopsin involved in proton pumping. Proc Natl Acad Sci U S A 88, 473-477 (1991). 4. Zimanyi, L. & Lanyi, J.K. Deriving the intermediate spectra and photocycle kinetics from time- resolved difference spectra of bacteriorhodopsin. The simpler case of the recombinant D96N protein. Biophys J 64, 240-251 (1993). 5. Otwinowski, Z. & Minor, W. Processing of X-Ray Diffraction Data Collection in Oscillation Mode. Methods Enzymol 276, 307-326 (1997). 6. Collaborative Computational Project, No.4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50, 760-763 (1994). 7. Brunger, A.T. et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D 54, 905-921 (1998). 7 8. Kleywegt, G.J. & Jones, T.A. xdlMAPMAN and xdlDATMAN-Programs for reformating, analysis and manipulation of biomacromolecular electron-density maps and reflection data set. Acta Crystallogr D 52, 826-828 (1996). 9. Yeates, T.O. Detecting and overcoming crystal twinning. Methods Enzymol 276, 344358 (1997). 10. Luecke, H., Richter, H.T. & Lanyi, J.K. Proton transfer pathways in bacteriorhodopsin at 2.3 angstrom resolution. Science 280, 1934-1937 (1998). 11. Adams, P.D., Pannu, N.S., Read, R.J. & Brunger, A.T. Extending the limits of molecular replacement through combined simulated annealing and maximum-likelihood refinement. Acta Crystallogr D Biol Crystallogr 55 ( Pt 1), 181-190 (1999). 12. Jones, T.A., Zou, J.Y., Cowan, S.W. & Kjeldgaard, M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47, 110-119 (1991). 13. Genick, U.K. et al. Structure of a protein photocycle intermediate by millisecond timeresolved crystallography. Science 275, 1471-1475 (1997). 14. Bhat, T.N. & Cohen, G.H. OMITMAP: An electron density map suitable for the examination of errors in a macromolecular model. J Appl Cryst 17, 244-248 (1984). 15. Evans, S.V. SETOR: hardware-lighted three-dimensional solid model representations of macromolecules. J Mol Graph 11, 134-138 (1993). 8