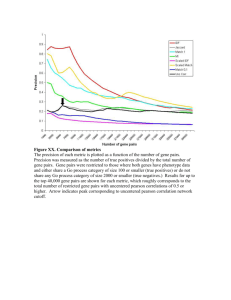
Directed scenarios of gene histories over an evolutionary
advertisement

Directed scenarios of gene histories over
an evolutionary tree as virtual
reticulation events
Boris Mirkin
School of Computer Science and Information Systems,
Birkbeck University of London, UK
and: E. Koonin, M. Galperin, Y. Wolf, T. Fenner
Outline:
- Three aspects:
Classification
Visualisation
Evolution
- What is a gene history
- Two approaches to building gene
histories:
- Mapping gene trees to species tree:
beautiful theory, poor results
- Mapping data straightforwardly:
Building static scenarios
Building directed scenarios
Classification:
Finding cluster structures
Interpreting cluster structures
Visualisation:
A cognitive activity of building a mental
image of a phenomenon in question for
gaining insight and understanding
(R. Spence, Information
Addison-Wesley, 2001)
Visualization,
S. Dali: Dream Caused by the Flight of
a Bee Around a Pomegranate One
Second before Waking Up (1944)
Our framework for visualisation:
Mapping data onto a known ground image
such as coordinate plane, geography
map, or a genealogy tree for gaining
insight and understanding
Ground image: An evolutionary (binary
rooted) tree, an outdated and simplified
schema, to be annotated with mapped
data
Task: Mapping data of gene families onto an
evolutionary tree to gain insight of gene
histories involving events of:
Emergence
inheritance
duplication
| two mechanisms of
horizontal transfer | evolutionary change
loss
Hybridization is not considered here
Data: 66 species
4873 gene families (COGs)
Ground image:
Evolutionary tree over 66 genomes
Example of a gene family:
COG1035
Coenzyme F420-reducing hydrogenase, beta
subunit
22 genes that occur in 8 species of 66
Distances between 8 genomes according to
COG1035 proteins
Afu
Mac
Mja
Mka
Mth
Nos
Sme
Syn
2.00
2.09
1.93
2.17
3.28
3.53
3.33
1.04
1.09
0.96
2.71
3.21
2.76
0.63
0.45
2.62
3.20
2.66
0.86
2.70
3.04
2.75
2.70
2.73
2.84
4.00
0.22
4.00
A history of COG1035 with mapping:
COG1035
66
129
128
65
64
63
130
62
61
125
127
60
124
122
121
123
126
120
118
131
119
59
58
57
56
55
54
53
117
116
52
51
50
49
48
111
47
110
115
46
109
112
108
107
45
44
43
42
41
114
40
103
39
102
100
99
101
38
37
36
104
98
35
97
96
34
33
32
113
95
105
31
94
93
91
92
90
30
29
28
27
26
25
24
23
86
22
85
21
82
84
106
20
81
19
80
87
83
18
79
78
77
17
16
15
14
13
76
88
75
12
11
10
73
89
71
72
74
70
69
9
8
7
6
5
4
3
68
67
2
1
Ecu
Sce
Spo
Hbs
Mac
Afu
Mka
Mth
Mja
Pho
Pab
Tac
Tvo
Sso
Ape
Pya
Tma
Aae
Dra
Cgl
Mle
Mtu
MtC
Syn
Nos
Fnu
Cac
Lla
Spy
Spn
Sau
Lin
Bsu
Bha
Mpu
Uur
Mpn
Mge
Ctr
Cpn
Tpa
Bbu
Xfa
Pae
Vch
Buc
Ype
Sty
Eco
EcZ
Ecs
Hin
Pmu
Rso
Nme
NmA
Ccr
Atu
Sme
Bme
Mlo
Rpr
Rco
Cje
Hpy
jHp
Two ways:
(i)
Building a within COG species tree
with the distances and then
MAPPING the gene tree to the
species tree;
models:
emergence
inheritance
duplication
loss
NO HORIZONTAL TRANSFER
(ii)
Mapping COG data onto the species
tree directly, never done before –
work in progress; models:
emergence
inheritance
horizontal transfer
loss
NO DUPLICATION so far
(i) Mapping individual gene
trees to a species tree
Reconciled tree
.
a
b
c
(i)
a1 b1
c1
a
(ii)
a2
b
c
b2 c2
(iii)
Figure 1. A gene tree (i) versus species tree (ii): the
reconciled tree (iii) explains the difference by the
duplication of the species tree with losses of lineages
leading to c1, a2 and b2.
(1) intuitively appealing pictures
(2) accommodates the presence of the so-called
paralogs, similar proteins in a species
(3) difficult to handle when there are many different
gene trees
Annotating duplication
+–
+ –
a
b
(i)
c
a
+
(ii)
b
+
c
–
Figure 2. Species in the left and right subtrees
of gene tree (i) annotated by symbols + and –
in the species tree (ii), with the follow-up
raising the symbols along the tree.
(1) less intuitively appealing
(2) maps different gene trees onto the same species
tree without changing the latter
(3) can be analysed mathematically:
Theorem: The total number of losses at mapping a
gene tree to the species tree is equal to the number of
incompatible pairs plus twice the number of
duplications [MIR 95], [EUL 98].
Lca mapping
a
b
(i)
c
a
+
b
+
(ii)
c
–
Figure 3. Lca mapping: internal nodes
of gene tree (i) onto species tree (ii):
1 contraction, 3 retractions.
(i) can be done in linear time over number of species
(ii) Theorem: The inconsistencies, contraction (1)
and retraction (2) in mapping parent-child pairs,
correspond to duplications and to those duplications
for which the collateral children are losses [EUL 98].
(iii) Theorem: The total number of losses is the
number of all the intermediate nodes in retractions
plus the number one-sided contractions under the lca
mapping M [EUL 98].
Distance matrices to model horizontal
transfer events: 2 stages
1 stage. Parsimoniously mapping an individual
gene set to the tree: algorithm PARS
PARENT
Inherited
Not inherited
CHILD1 Gain Loss
Inherited Gi1 Li1
Not inher Gn1 Ln1
Gain Loss
Gi
Li
Gn
Ln
CHILD2 Gain Loss
Inherited Gi2 Li2
Not inher Gn2 Ln2
Figure 4. Events in a parent-children triple
according to a parsimonious scenario.
Initial setting: At a leaf node the four sets Gi,
Li, Gn and Ln are empty, except that
Gn ={a} if gene a is present in the given leaf or
Li = {a} if a is not present.
Criterion:
#LOSS + gp#HT
MIN
Presence-absence pattern of COG0572
Uridine Kinase
y o
z
m k a
p
b l
w d c r n
s
f
g
h e x
j
u t
i q
v
Reconstructed MP history of COG 0572
Uridine Kinase (A static scenario)
presence
gain
6 events:
4 losses
2 gains
inheritance
loss
y
o
z
m k a p b l w d
c r
n
s
f g h e x
j
u
t i
q
v
Reconstructing LUCA at 26 cellular species
- 26 species (bacteria, archaea and yeast)
- 3166 gene families (COGs)
- gain penalty from 0.1 to 10
Reconstructed contents of the root (LUCA) an external criterion for selecting gain penalty:
At gp=1, 572 genes in LUCA
best approximated an organism
capable to survive [Mir03]
This leads to equal empirical
frequencies of losses and gains
2 stage. Principle of maximum correlation
(i) evolutionary tree must be clocked
(ii) directed scenario: Each gain, g, has its
source, s
L
L
S
S
T
R
T
R
Time
100
70
20
a
b
c
d
e
a
b
c
d
e
Figure 5. Horizontal transfer event; (C1) directly
from node S to node a and (C2) from the middle of
T's life-span to the middle of a's life span.
Distances between a, b and c:
No transfer
C1
c d
D=|200 200|
|
140|
C2
c d
c d
D1=|70 70| D2=|100 50| a
| 140|
|
140| c
Principle:
find such a directed scenario that
is best matching the observed distance matrix
between proteins
Virtually reticulated trees?
b a c d
e
b
cad
e
Figure 6. Changed tree topologies corresponding to directed
scenarios C1 and C2 on Figure 5.
Synchronization of source-target timing
D
B
D
D
C
B
C
A
B
C
A
A
Figure 6. Generic patterns of interrelation
between life spans of a horizontal transfer
target, AB, (vertical line on the left) and its
source, CD (vertical line on the right), to
generate the synchronising distance between
them, AB at (i) or AD at (ii) and (iii).
Deriving
weight:
horizontal
transfer
penalty
At 66 genomes, with principle of maximum
correlation applied to build correlated
distances over maximum parsimony scenarios,
of 4872 COGs:
3975 correspond to
gp=1
602
to
gp=2
295
to
gp=3
thus concurring with the results of our
previous analysis of 26 genomes
Future work:
- Complete a mathematical
computational framework for
principle of maximum correlation
and
the
- Include paralogous (representing
multiple copies of a gene within a
genome) proteins to model duplication
- Extend model to virus and other
species types (to include host effects)
- Include more data on gene families