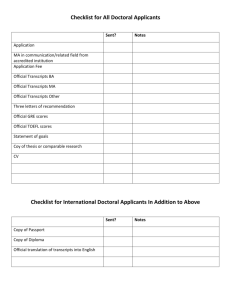
Excel spreadsheet
advertisement

Explanatory text for supplementary data, “How self-tolerance prevents B cell mitogenesis: contrasting effects of FK506 provide a rationale for improved immunosuppressants”. Corresponding author: Chris Goodnow Columns A and B: probeset ID, accession Probeset ID: An identifier to describe the collection of probe pairs, either for controls or transcripts. Transcripts are usually designated with the unigene cluster number (at the time of chip design). As these numbers are sometimes retired or updated in the light of new seqeuncing data, an exemplar accession number from each cluster is provided under accession. Recent Entrez results are provided in the “Entrez hits” folder. Columns C-BA: Average difference intensities (a trimmed average of the perfect matchmismatch values for the approximately 20 probe pairs tiled for each gene. B cell treatments are coded as follows: Rest: mock stimulated by incubation at 37oC Mu: stimulated with goat anti-mu (Jackson Labs) at 10 g/ml HEL: stimulated with Hen Egg Lysozyme (Sigma) at 500 ng/ml FK: preincubated for 15’, 37oC with FK506 at 10 ng/ml and then stimulated in the continued presence of FK506 with either HEL or Mu. PD: preincubated for 45’, 37oC with PD98059 (NEB) at 10 or 20 m (10uMPD and 20uMPD respectively) and then stimulated in the continued presence of PD98059 with either HEL or Mu EGTA: stimulated with either HEL or Mu in the presence of EGTA at 3 mM Ionomycin stimulated with ionomycin at 1 m Stimulations were for 1 hour unless noted as 6 hr. The experiment number is given after the letter “X”. Experiments 1-10 were performed on B cells purified by negative depletion of CD4, CD8 or Mac-1 positive staining splenocytes. Experiments 11 and 12 were carried out with naïve and tolerant B cells purified by FACS sorting. Non-transgenic B cell from B6 mice were used in experiments 1-4, transgenic IgHEL B cells were used in experiments 5-12. Column BC-BF: p(unchanged at 1 hr) t test, Npos 1 hr, Nneg 1 hr, median fold change For each probe pair, the PM-MM values were compared by paired t test between resting and stimulated samples in experiments 1-7. Positive t values associated with a probability of less than 0.1 were scored as increased, negative t values with a probability of less than 0.1 were scored as decreased. The number of increased and decreased probe pairs associated with each gene was determined and called npos 1 hr and nneg 1 hr respectively (of the approximately 20 probe pairs tiled for each gene). The distribution of npos and nneg is binomial where p=0.1 and n=number of probe pairs for that gene. The probability of scoring npos or more of the total number of probe pairs was determined. The same analysis was done for decreased genes using nneg. The resulting probability is recorded in P(unchanged at 1 hr) t test. Median fold change was calculated as the median stimulated/resting ratio of average difference intensities over the seven experiments. Average difference intensities less than 5 were considered indistiguishable from 5 for this calculation. 280 transcripts had median fold change greater than 1.75 or less than 1/1.75. Of these 280 transcripts, a significant change was determined if the probability of the transcript being unchanged (column BA) was less than 0.00018. This ensures a probability of less than 5% of one or more false positives in 280 trials. Columns BG-BH: inc 1 hr, dec 1 hr A transcript was scored as increased if median fold change (column BE) was greater than 1.75 and if the P(unchanged at 1 hr) t test (column BB) was less than 0.00018. These transcripts are marked with 1 in inc 1 hr (column BF). A transcript was scored as decreased if median fold change (column BE) was less than 1/1.75 (0.57) and if the P(unchanged at 1 hr) t test (column BB) was less than 0.00018. These transcripts are marked with 1 in dec 1 hr (column BG). Columns BJ-BL: average fold change 6hr, inc 6 hr, dec 6 hr Average fold change is calculate as average of HEL6hrX5/restX5 and HEL6hrX7/rest6hrX7. Average difference intensities less than 5 were considered indistiguishable from 5 for this calculation. A transcript was considered increased at 6 hours if the data satisifed the following queries. For a given transcript, each probe pair was scored as increased in sample A relative to sample B if (PM-MM)A-(PM-MM)B > 30 and (PM-MM)A >1.3x(PM-MM)B and decreased by the reverse. A gene was scored as increased in each pairwise comparison if the number of positive probe pairs was 3 or greater, the ratio of positive to negative probe pairs was greater than 3 and the average difference intensity of the stimulated sample was moe than 1.8 fold greater than the resting sample. Decreased genes were determined by the reverse of this algorithm. Samples that were compared with this algorithm were HEL6hrX5 with restX5 and HEL6hrX7 with rest6hrX7. Transcripts scored as increased in both comparisons are marked as “2” in inc 6 hr, transcripts scored as increased in only one comparison are marked as “1” and in neither comparison as “0”. Likewise for decreased transcripts in dec 6 hr. Based on comparisons of all genes between closely matched samples the false positive rate of this query was empirically determined to be approximately 0.055 per gene in any pairwise comparison. Consistent changes across the 2 experiments have a false positive rate of 0.003 (=0.0552). Column BN: median % induction in presence of FK506 1 hr Calculated as median ((antigen/FK506-mock)/(antigen-mock))x100 over 5 experiments of antigen receptor stimulation for 1 hr in the presence or absence of FK506. Columns BP-BW: p(unchanged in tolerance) t test neg depletion, Npos tolerance, Nneg tolerance, inc sorted tolerance, dec sorted tolerance, inc tolerance, dec tolerance, median fold change tolerance For each probe pair, the PM-MM values were compared by unpaired t test between tolerance and naive in experiments 8, 9 and 10. Positive t values associated with a probability of less than 0.1 were scored as increased, negative t values with a probability of less than 0.1 were scored as decreased. The number of increased and decreased probe pairs associated with each gene was determined and called Npos tolerance Nneg tolerance respectively (of the approximately 20 probe pairs tiled for each gene). The distribution of npos and nneg is binomial where p=0.1 and n=number of probe pairs for that gene. The probability of scoring npos or more of the total number of probe pairs was determined. The same analysis was done for decreased genes using nneg. The resulting probability is recorded in p(unchanged in tolerance) neg depletion. Two comparisons of sorted naïve and tolerant B cells from experiments 11 and 12 were performed as for the 6 hr comparisons. Transcripts scored as increased in both comparisons are marked as “2” in inc sorted tolerance, transcripts scored as increased in only one comparison are marked as “1” and in neither comparison as “0”. Likewise for decreased transcripts in dec sorted tolerance. 149 transcripts were either (i) at least 1.75 fold upregulated in tolerant compared to naïve B cells and upregulated in at least one of the FACS sorting experiments or (ii) at least 1.75 fold downregulated in tolerant compared to naïve B cells and downregulated in at least one of the FACS sorting experiments. Of these 149 transcripts, a significant change was determined if the probability of the transcript being unchanged (column BN) was less than 0.00034. This ensures a probability of less than 5% of one or more false positives in 149 trials. Upregulated transcripts are marked with 1 in inc tolerance (column BU), downregulated transcripts are marked with a 1 in dec tolerance (column BV) Median fold change tolerance (column BW) was calculated as the median tolerance/naive ratio of average difference intensities for 6 comparisons, including two experiments using cells purified by FACS sorting. Average difference intensities less than 5 were considered indistiguishable from 5 for this calculation. Columns BY-CE: gene number (column BY) is a unique identifier provided for sorting the spreadsheet. Chip (column BZ) refers to the array (of the set of four) on which the gene was tiled. Gene name (column CA) is a common gene name used in the figures and column CB contains the Entrez definition for the exemplar accession number as supplied by Affymetrix. Entrez definition lines for a recent (9/12/99) batched Entrez search are given in the spreadsheets blank entrez hits, EST entrez hits and gene entrez hits in the “Entrez hits” folder. Genes, ESTs, controls and entries in which no Entrez definition was provided are designated in gene/EST (column CC). Entries that had no match in the database for the exemplar accession number are marked with a * in withdrawn (column CD). Columns CF-CJ: identification of transcripts that are affected by the presence of the IgHEL transgene. P(IgHEL vs B6) is the probability that a gene was altered in expression pattern between IgHEL and B6 B cell preparations. Fold change IgHEL vs B6 is the fold change between these cell preparations and transcripts that had a 1.8 fold change or greater with p<0.0001 are shown in IgHEL vs B6, >1.8* change, p<0.0001. Only one transcript, that for the IgHEL transgene itself, passed this query. P(Inter-action IgHEL* stimulation) is the probability that IgHEL transgenic cells responded differently to HEL than B6 cells responded to anti-mu. Transcripts which are altered by 1 hour antigen stimulation that showed a significant interaction with IgHEL are shown in Inter-action IgHEL* stimulation. Only MIP-1b passed this query. Columns CL-CQ: identification of transcripts affected by in vitro incubation. P(incubation) is the probability that in vitro incubation affected the expression level. Average expression levels for IgHEL cells that were or were not incubated in vitro and the standard deviations are shown in avg no incubation, avg incubation, stdev no incubation, stdev incubation. Genes that are affected by incubation in vitro are shown in Effect of incubation, (greater than 1.8 fold change and p<4e-5). Columns CS-CU: identification of transcripts that appeared altered between tolerant and naïve cells in the negative depletion samples but were not confirmed in samples purified by FACS. Transcripts that were significantly altered between cell preparations purified by negative depletion from IgHEL and IgHEL: sHEL mice are defined as median fold change tolerance neg dep only greater than 1.75 and p(unchanged in tolerance) t test, neg depletion (column BP) < 5.9e-5. 57 tilings, representing 56 genes, passed this query and are shown in change tolerance neg dep data only. 18 of these genes were affected by purification method in that there was a significant interaction effect between method of purification (FACS or negative depletion) and cell phenotype (tolerant or naïve), marked in change tolerance neg dep only & affected by purification. The explanation for many of these interactions was a depletion of contaminating myeloid and erythroid transcripts in the FACS purified samples relative to the samples purified by negative depletion. This is presented in supplementary figures 1c and d. Column CX-CY: of activation response, what is conserved in tolerance? These 16 genes were part of the activation response at 1 or 6 hours (1.75 fold change, p<0.00018 for 1 hour; changed in 2 of 2 experiments for 6 hours) and were regulated in the same direction as part of the tolerance response (1.8 fold change, changed in tolerance in at least one of two FACS experiments, p<0.05). of tolerance response, what is conserved in activation? These 18 transcripts were part of the tolerance response by stringent criteria (p<0.00034) and had some evidence for being regulated in the same direction by foreign antigen (either median fold change at 1 hour greater than 1.8 and p<0.05 or changed in 1 of 2 experiments after 6 hours stimulation).