DNA SEQUENCING Objectives: 1) To understand these strategies of
 To understand these strategies of")
DNA SEQUENCING
Objectives:
1) To understand these strategies of dideoxy DNA sequencing. a) Shotgun b) Primer walking c) Single stranded, vs. double stranded sequencing. d) Cycle sequencing e) Automated sequencing
2) To understand the process of data assembly.
3) To recognize various patterns reflecting errors in base calling.
Why sequence?
Knowledge of the DNA sequence is required for most kinds of studies that are carried out with
DNA. These include footprinting, primer extension, RNAse protection to distinguish closely related genes, in vitro mutagenesis, analysis of mutants or polymorphisms (by PCR), construction of recombinant genes, over expression of the gene in E. coli, and analysis of splicing patterns. Moreover, DNA sequencing has replaced other methods to achieve the most basic characterization: 1) sequencing has virtually replaced all restriction mapping on a resolution of less than 1 kb, 2) sequencing is essential in distinguishing the members of a multi gene family, 3) comparison of DNA sequence to peptide sequences is the most common and reliable method of establishing the identity of the gene. More and more often, comparison of the sequence to other known sequences is used to infer aspects of the function of a new gene.
General review of dideoxy sequencing
Dideoxy sequencing requires enough forehand knowledge of the sequence to make a primer. The products can be labelled by end-labeling the primer, or by incorporating radiolabel during the extension.
The procedure requires a denaturing gel that can resolve the products with one base resolution. This is done on a 4-8% polyacrylamide gel containing 7-8 M urea. One can reasonably expect to get 250-300 bases of sequence from each ladder, and to extend the sequence out to 500-600 bases from the primer by running the gel longer.
1
The above described method based on slab gels and autoradiography have mostly been replaced by fluorescent labeling and capillary electrophoresis. See below.
Strategies of dideoxy sequencing.
Primer walking
For sequencing a small (<2Kb) clone, one generally starts with primers complementary to the vector. Originally, inserts for sequencing were cloned into bacteriophage M13. M13 replicates in the cell as a double stranded circle (called RF for replicative form), and then switches to a single stranded mode of replication that produces a single stranded circle for packaging into the phage head. DNA recovered from the phage particle is single stranded (called + strand) and can be used as a single stranded template for sequencing.
M13 does not lyse or kill the host, and so it can be propagated as a plasmid. However, this is an undesirable way to propagate an M13 clone because you inadvertently select for deletions that remove the phage genes, and usually the insert too. Consequently, M13 clones must be grown infectively to maintain selection for the phage genes. This is done by inoculating a large amount of bacteria with a small amount of phage and allowing the phage to overtake the bacteria during a short period of growth. A phage stock is then saved to start future cultures.
Phagemids
Phagemids are plasmids that contain the M13 single stranded origin of replication ( ori ). (The closely related phage, f1, is often the source of the ori ). Phagemids also have a normal plasmid ori , and a drug resistance marker; and are constructed, transformed into bacteria, and grown like normal plasmids.
Alternatively, by coinfecting with a helper M13 phage, one can cause the phagemid to be replicated to a single stranded circle and packaged into M13 phage heads. The helper phage is genetically crippled so that it does not make its own DNA.
Satisfactory methods are now available for priming directly on a denatured double stranded plasmid. The double stranded methods are more exacting, because of the risk of the plasmid renaturing and displacing the primer. However, most people currently prefer the double stranded methods, because you can get sequence from both strands from the same template. Usually sequencing from double stranded templates is done by thermal cycling (see below), where the concentration of template can be kept very low and problems due to template renaturation are thus avoided.
2
In primer walking, the sequence gained from priming with the vector primer is then used to custom design a new primer, and one walks stepwise through the sequence in this fashion.
A problem with this strategy is that a significant fraction of custom synthesized primers inadvertantly match a second site in the clone well enough to produce a double ladder, which is unreadable. This problem can be largely overcome by prescreening all prospective primer sequences against the vector sequence, and avoiding primers that have greater than 6 perfect matches at their 3' ends.
With thermal cycling, mispriming is subtantially suppressed by priming at high temperature.
Shotgun Sequencing
For larger sequencing projects, generally some form of shotgun cloning is employed. The DNA to be sequenced is broken into a large collection of semi-random fragments, either by cutting with frequently cutting restriction enzymes, or by shearing (for example, with a sonicator). The fragments are cloned and each is sequenced from a vector primer without any prior characterization. A computer program searches the collection of sequences for overlaps, and assembles a contiguous sequence from the fragmentary sequence data. The problem with this approach is that near the end of the project, one has a greater chance of retrieving another sequence from a region that is already done, than of gaining a sequence that closes the last gaps. Generally, one should expect to sequence 8 times the total sequence length to fill the last gaps. Most groups gather data by shotgun cloning until they reach a point of diminishing return; then they switch to a primer walking strategy.
3
Directed Deletions
Several strategies exist to create a set of nested deletions from one end of a cloned insert. The purpose is to place the vector priming site next to a variety of internal positions to permit sequencing all the way through the insert with the same primer. One strategy is shown below that uses an enzyme named
Bal31 that chews in from the end of double stranded fragments leaving a blunt end.
BE stands for "blunt ended restriction site" in the above figure. One can arrange for a uniformly spaced set of deletions by controlling the extent of digestion, sizing the excised insert, or sizing the recloned plasmids.
Cycle sequencing
In cycle sequencing, the extension is done with a thermo-stable DNA polymerase at high temperature. A major advantage of this is that the primer can be annealed at high stringency, thus avoiding adventitious priming at alternate sites. It is then possible to sequence directly off of lambda, or
4
even P1 templates. A second advantage is that the reaction can be conducted in a thermal cycling machine (PCR machine) and cycled from a stringent temperature for annealing and polymerization, up to a higher temperature to denature the product from the template, and back to annealing temperature to reinitiate another round of priming and synthesis. In this way, product can be accumulated from relatively small amounts of template. (Note: this is not a chain reaction, because only one primer is used, and the product does not itself become a template in later cycles. Therefore, product accumulates proportionately to the number of cycles; not exponentially.)
If done with radioactivity, this method is usually used with end labeled primers, since the methods to suppress pausing during continuous labeling are difficult to reconcile with the cycling strategy. More commonly, it is done with fluorescent labeling. In particular, if the fluorescent label in put on the dideoxy terminator, then pausing becomes irrelevant. This also allows use of ordinary unlabeled oligo primers.
However, the method now becomes dependent on purifying away other oligos (most notably PCR primers).
Automated sequencing
Automated sequencing is conducted with fluorescent labeling rather than radioactive labeling. In the original application, four reactions are conducted with four different primers, each with a different color of fluorescent dye attached. All four products are run in the same lane on a special machine. The gel is run continuously past a fluorescence sensor at a fixed position down the gel. The sensor automatically determines the color of each passing peak and outputs the corresponding DNA sequence to a computer.
An improved alternative, designed to avoid having to fluorescently label a wide variety of custom synthesized primers, is to attach the fluorescent labels to the dideoxy terminators.
Modern DNA automated sequencing is mostly done on an capillary gel apparatus using fluorescently labeled dideoxy terminators. The four different terminators are labeled with different color dyes, so the reaction can be done in one tube. The detector attached to the sequencer can distinguish the four colors.
The data is output as a four-color chromatogram (often called a "trace"). See the html supplement on the course web page for comparable information to this chapter illustrated with four color chromatograms.
Automated capillary sequencers generally get about 500 nt of high quality data per trace, and up to another
400 nt of data with increasing loss of resolution.
Capillary sequencers are loaded by a process called "electrokinetic injection", which means by electrophoresis. Salt will interfere with the loading, so there is usually salt clean up step after the thermocycling. This can be as simple as ethanol precipitation, or may involve a resin. This step also removes the unincorporated dye-terminators, including some extraneous low molecular weight fluoresent compounds in the reaction mix. Failure to remove these completely causes a phenomena called "dye blobs" for lack of a better term. A "dye blob" is a large peak of a single color covering several nucleotides in the four color chromatogram. The base caller will frequently call a run of the base corresponding to that color rather than the true sequence.
5
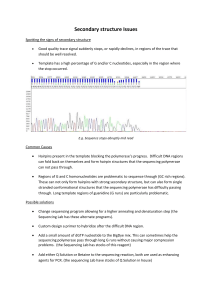
Problems in data acquisition
Compression
Compression is caused by secondary structure in the sequencing reaction products while they run on the sequencing gel. Compression is apparent as a zone where a number of bands are compressed into a small space, above which is a region with fewer bands than should be expected from the spacing. The order of the bands in the compressed zone may not reflect the true sequence. Compression has absolutely nothing to do with the sequencing reaction. The claim of polymerase vendors that purchase of their product will eliminate compression is a misstatement of the pausing problem to be discussed below.
Compression reflects inadequate denaturing conditions in the gel. People often fail to understand that the heat output of the gel is part of the denaturing conditions. Running the gel at too low a wattage will increase the number of compressions. Compressions nearly always involve runs of C's and G's, therefore they are more common in G+C rich sequence.
Compressions are dealt with by using analogues of dGTP that form weaker base pairs. Commonly used analogues are inosine, and 7-deaza guanine.
Pausing
The DNA polymerase will either stop or pause on templates that have any of the following: strong secondary structure, a complementary strand annealed in the way, or a natural termination signal for the polymerase. The polymerase may also pause frequently if the concentration of one or more nucleoside triphosphates is too low, the polymerase is dying, or reaction conditions are otherwise suboptimal.
Pausing is recognized by a pattern of bands across all four lanes with no disruption in the spacing, or just loss of signal if the sequencing is by terminator labeling.
Pausing can generally be eliminated by changing the polymerase, or by correcting the deficiency in the conditions. Pausing is particularly a problem when trying to incorporate label during the extension, because the chemical concentration of the labeled triphosphate is usually too low to support efficient polymerization. This is usually dealt with by first labeling with a short extension of just a few bases, and then following with the termination mixtures including a high concentration of all four dNTPs. Note that
6
this doesn't work for cycle sequencing because there's no way to repetitively cycle between the two different extensions. If one must use radiolabel during extension in cycle sequencing, then it is imperative to keep the concentration of the labeled base up even at the expense of specific activity.
One solution to the pausing problem that can work with cycle sequencing is to follow with an
(untemplated) terminal transferase reaction to try to extend all non terminated fragments to a length that takes them out of the way of readable data (Fawcett and Bartlett, BioTechniques 9:46-48 (1990)).
With fluorescently labeled thermo cycle sequening, the most common problem causing the polymerase to pause is a large inverted repeat that snaps back to make a double stranded region that blocks polymerase progress.
Band drop out.
A common problem is uneven labeling of bands such that the fainter bands drop out of the pattern.
This is particularly problematic when using some base analogues (inosine in particular) while trying to suppress compressions. It is caused by a process called phosphorolysis, whereby the polymerase reverses itself after adding a ddNMP and remove the terminator and try to extend the stand again. This reversal uses pyrophosphate that builds up during the sequencing reaction. The problem is very sensitive to the context of the sequence, and so only particular bands reproducibly drop out. It is possible to eliminate this problem by incorporating pyrophosphatase into the reaction. However, there isn't a thermo-stable pyrophosphatase for use in thermal cycle sequencing.
Template heterogeneity
When M13 replicates, its replication system stutters on homopolymer tracts (like poly(A)). This causes the template to be heterogeneous in the length of these tracts such that you can't sequence through them.
The problem is solved by transferring the insert to a plasmid vector.
Sequence assembly
Sequence autoradiogams can either be read manually, semiautomatically (for example, with a hand held pen on a digitizer pad), or fully automatically. Four color chromatograms are always read by an automated base caller. The algorithms for reading chromatograms are often sophisticated and have to be customized to the make of instrument for maximum capability. A good program will assign quality values to each base call, and a good sequence assembler will automatically take the quality values into account to avoid having bad data override good data in overlapping reads. Quality values are usually defined as 10/(probability of an error). So 10 means 10% chance of error, 20 means 1%, etc. The standard for the genome project is 50. Since even good single reads generally average about 20, the assembler has to use artificial intelligence to form a joint estimate of the quality value for the consensus of several reads. The best assemblers know to account for systematic errors in various kinds of data by giving greater weight to a call made by two different kinds of "chemistries" than to the same call made by the same reaction run twice. The most important way to change the "chemistry" is to sequence from the complementary strand.
7
There are basically two different strategies depending on the capabilities of the software. If the software is powerful enough to selectively ignore low quality data in assembly, then the data can be just streamed to the assembler without human intervention. Generally there should be an automated screen to mask vector sequences. For software that is easily confused by poor quality data embedded in the raw traces, one generally has to trim the readings down to the quality data prior to assembly. This can be done manually, or automatically by reference to called quality values. The former is time consuming, and in either case data that might accelerate overlapping contigs is lost by trimming. However, for small projects this is how it is usually done because of the greater learning curve for using the software with a higher component of artificial intelligence.
In all shotgun strategies, it is necessary to pay attention to how the software deals with chimeric clones in shotgun strategies. A common problem is for falsely contiguous sequence to establish the order of assembly early, and for the assembler to subsequently throw out or misassemble correct data because it disagrees with the earlier data.
The presence of repeated sequences within the region being assembled may require manual intervention. In severe cases, you may have to do some restriction mapping to resolve the true order of the sequences.
For example, in the figure above, the two black boxes are a perfect inverted repeat. You have no way to know from the gel readings themselves whether 1 overlaps 2 and 4 overlaps 5 as shown, or if 1 overlaps 4 and 5 overlaps 2 inverting the central segment. You could resolve this by sizing the double digestion fragments produced by restriction enzymes A+B and B+C. The sequence data would reveal suitable enzymes to use for this purpose.
8