Corresponding author: Jack Smith, jsmith@email
advertisement

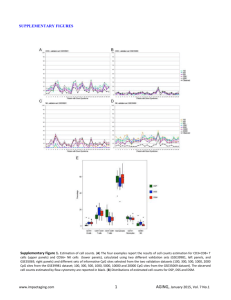
Corresponding author: Silvia Pineda Sanjuán, spineda@cnio.es Please, indicate the type of presentation you prefer (oral or poster): Oral presentation Please, choose the two main topics of your contribution from the following list. Primary topic: Statistical Genetics Secondary topic: Bioinformatics 1 Statistical approaches for the integration of ‘omics’ and epidemiological data: an application to bladder cancer. 1,2 Silvia Pineda-Sanjuán , Roger Milne1, Kristel Van Steen2, Núria Malats1 1 Spanish National Cancer Research Center (CNIO), Madrid, Spain; 2University of Liege, Belgium Abstract Integrating different ‘omics’ datasets may give us a new view of the biological mechanisms involved in disease. Advanced methods will be necessary to deal with high-volume, multidimensional data. We propose a three-step process to integrate data from the genome, transcriptome, and methylome, together with epidemiological risk factors for bladder cancer (BC). Here we present the results from the first step of the process. Keywords: omics, integration, statistics. Introduction: Many data are becoming available in the context of ‘omics’ studies (i.e., genomics, transciptomics, epigenomics) but computational problems arise in analysing them because of the large number of parameters (‘p’) and relatively small number of observations (‘n’). Another problem is the heterogeneous nature of the data. Sometimes, it is not possible to extract enough information from a single ‘omics’ dataset to understand the underlying biological mechanisms. New methods are therefore needed to integrate multiple different datasets. An additional challenge lies in ensuring that the results from an integration analysis are interpretable. Other statistical problems include data overfitting and multicollinearity [1]. Recently, the idea of data integration has become very important in ‘omics’ research and many articles have been published in this context [2-5]. Different statistical methods need to be integrated and new approaches need to be adapted to the emerging ‘omics’ data in order to obtain greater precision, accuracy and statistical power [6-10]. In the present study we consider an integration of different ‘omics’ data in bladder cancer cases, comprising common genetic variation (1M-SNP), in blood and tumor, and tumor DNA methylation and gene expression, together with epidemiological information, in a way that appropriately represents the relationship between the five types of data. Material and methods: Patients (N=70) recruited in the pilot EPICURO study with available fresh tissue were considered in this study. All of them were histological confirmed cancer cases recruited in 2 hospitals in Spain during 1997-1998. Not all individuals provided data for the different ‘omics’ data. This data comprise transcriptomics (Affymetrix DNA Microarray Human Gene 1.0 ST Array), epigenomics (Infinum Human Methylation 27 BeadChip Kit), and blood and tumor tissue genomics (Illumina 1Million SNP-array). The analytical plan considers a three-step process: (1) Comparison of 1M-SNP in blood & 1M-SNP in tumor tissue to identify regions with high rates of somatic changes. To do this we calculated the percentage of agreement and the weighted kappa measure to take into account that a change from common homozygote to heterozygote was not the same a change to rare homozygote. We also compared DNA Cytosine-phospate-Group (CpG) sites and tumoral gene expression probes using the Spearman correlation for non-normally distributed variables. (2) Assessment of the association between 1M-SNP (tumor tissue and blood separately) and each of expression and methylation datasets: on a “1-to-1” basis using ANOVA or Kruskal Wallis, depending on the distribution of the data [11]; on a “1 to n” basis using conditional inference trees [12] and penalized regression [13] ; on an “n to m” basis using canonical correlation analysis. (3) Integration of the ‘omics’ datasets in a ‘network’ together with epidemiologic variables such as age, gender and smoking status. Results: After Quality Control (QC), genotypes were available for 1,037,880 SNPs in blood and tumor DNA from 39 and 46 patients, respectively, and in both tissues for 16 patients. Gene expression was determined for 21,254 annotated probes in 37 patients, and DNA methylation measured at 26,617 CpG XIV Conferencia Española de Biometría 22 a 24 de mayo de 2013 CEB2013 Ciudad Real 2 sites for 54 patients. Methylation probes were classified into three categories: CpG island, CpG island shore (sequence up to 2kb from an island) and outside CpG island/shore. The number of comparisons we performed between expression and methylation was 860,288,057, based on data from 30 patients with both measures. Expression-methylation probe pairs were classified into three possible effects: cis-acting if there was at most 500kb between the probes; trans-acting if they were on the same chromosome but more than 500kb apart; and trans-acting-outside the chromosome they were on different chromosomes. For the comparison between genotypes in blood and tumor, we found some difficulties interpreting the weighted kappa in those cases were the probability by chance is higher than the observed proportion of agreement given a number of 14,385 SNPs with kappa ≤ 0. Nevertheless with both measures (kappa and agreement percentage) we found similar results. We identified that in chromosome 9 the percentage of agreement and the kappa was systematically lower than in the rest of the genome (Figure 1). For the comparisons between expression and methylation levels, we obtained 27,964 strongnegative (ρ < -0.7) and 104,748 strong-positive (ρ > 0.7) associations between gene expression and methylation. Of the methylation probes in these associations, 97,852 were CpG island, 21,205 CpG shore and 13,655 outside of a CpG island/shore. There were 182 cis-acting correlations, 7,216 trans-acting correlations and 116,459 trans-acting outside chromosome. For a total of 8,855 we were not able to annotate the gene at probe level. Results are shown in table 1. For those who were inside a CpG island in a cis-acting relationship, we expected a negative correlation (40 out of 104), but we also found a positive one (64 out of 104) Conclusions: Here we present preliminary results from an ‘omics’ integration approach in bladder cancer. We observed some regions with high percentage of somatic mutations, especially in chromosome 9, usually deleted in BC. We have also begun to describe the complexity of the relationships between methylation and gene expression that will help in the implementation of the next steps. XIV Conferencia Española de Biometría 22 a 24 de mayo de 2013 CEB2013 Ciudad Real 3 Tables and figures Figure 1: Kappa weighted measure (not p-values) from all chromosomes (SNPs=1,036,938) Cis-acting Trans-acting Trans-actingoutsideChromosome Negative Correlation Positive Correlation CpG island 40 64 CpG shore 13 20 CpG outside 10 35 CpG island 584 3,214 CpG shore 343 1,046 CpG outside 529 1,500 CpG island 9,461 54,799 CpG shore 4,423 16,750 CpG outside 7,496 23,530 Table 1: Cross table of the CpG position and the sign of the correlation by effect of the relation between expression and methylation. XIV Conferencia Española de Biometría 22 a 24 de mayo de 2013 CEB2013 Ciudad Real 4 Bibliography 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Hamid, J.S., et al., Data integration in genetics and genomics: methods and challenges. Hum Genomics Proteomics, 2009. 2009. Serizawa, R.R., et al., Integrated genetic and epigenetic analysis of bladder cancer reveals an additive diagnostic value of FGFR3 mutations and hypermethylation events. Int J Cancer. 129(1): p. 78-87. Greenawalt, D.M., et al., Integrating genetic association, genetics of gene expression, and single nucleotide polymorphism set analysis to identify susceptibility Loci for type 2 diabetes mellitus. Am J Epidemiol. 176(5): p. 423-30. Bell, J.T., et al., DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 12(1): p. R10. van Eijk, K.R., et al., Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics, 2012. 13: p. 636. Parkhomenko, E., D. Tritchler, and J. Beyene, Sparse canonical correlation analysis with application to genomic data integration. Stat Appl Genet Mol Biol, 2009. 8(1): p. Article 1. de Tayrac, M., et al., Simultaneous analysis of distinct Omics data sets with integration of biological knowledge: Multiple Factor Analysis approach. BMC Genomics, 2009. 10: p. 32. Palermo, G., P. Piraino, and H.D. Zucht, Performance of PLS regression coefficients in selecting variables for each response of a multivariate PLS for omics-type data. Adv Appl Bioinform Chem, 2009. 2: p. 57-70. Poisson, L.M., J.M. Taylor, and D. Ghosh, Integrative set enrichment testing for multiple omics platforms. BMC Bioinformatics. 12: p. 459. Mayer, C.D., J. Lorent, and G.W. Horgan, Exploratory analysis of multiple omics datasets using the adjusted RV coefficient. Stat Appl Genet Mol Biol. 10(1): p. Article 14. Szymczak, S., B.W. Igl, and A. Ziegler, Detecting SNP-expression associations: a comparison of mutual information and median test with standard statistical approaches. Stat Med, 2009. 28(29): p. 3581-96. Strobl, C., et al., Conditional variable importance for random forests. BMC Bioinformatics, 2008. 9: p. 307. Tibshirani, R., Regression Shrinkage and Selection via the Lasso. Journal of the Royal Statistical Society. Series B (Methodological), 1996. 58(1): p. 21. XIV Conferencia Española de Biometría 22 a 24 de mayo de 2013 CEB2013 Ciudad Real