Protease Inhibitors
advertisement

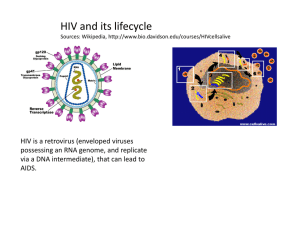
Tawit Suriyo How do the drugs (Reverse Transcriptase, Integrase and Protease Inhibitors) work? Content I n t r o d u c t i o n W h a t a r e H I V i n f e c t i o n a n d A I D S ? The step in the life c ycle of H IV that might be stopped. The types of anti-HIV drugs that we have now Reverse transcriptase inhibitors Integrase inhibitors Protease inhibitors Conclusion Introduction What are HIV infection and AIDS? Human Immunodeficiency Virus (HIV) depends on the cells that it infects to make new copies of itself. These new copies of HIV go on to infect other cells. In infected person, over 10 billion new copies of the virus can be made every day. One of HIV’s main targets is a white blood cell called T helper cell or CD-4 positive cell. These white blood cells are important because they trigger other infection-fighting cells to start working. HIV destroys CD-4 cells, and when the number of CD-4 cell drops to certain level, the body’s immune system weakens. As a result, organisms such as fungi, viruses and parasites can cause serious infections in people with HIV. When these infections occur, or when the number of CD-4 cells drops below a certain level, a person with HIV infection is said to acquire immunodeficienc y s yndrome (AIDS). (1) The step in the life cycle of HIV that might be stopped . 1. Attachment and entry of the Viral RNA : The life cycle of HIV begins when HIV attaches itself to the membrane of the target host cell. There are at least two receptors on T-lymphocytes to which the HIV binds. The primary receptor, called CD-4, and a second receptor, called a transmembrane receptor are critical for infection to occur. HIV infection of a lymphocyte requires attachment of the virus to the cell membrane through both of these ligand-receptor links. Tight attachment of the viral particle to receptors on the lymphocyte membrane enables fusion with the cell membrane. The viral contents, including viral RNA then empty into the cytoplasm of the host cell. 2. Reverse transcription : The step that converts viral RNA into DNA. Reverse transcriptase (RT) reads the sequence of viral RNA nucleic acids that have entered the host cell and transcribes the sequence into a complementary DNA sequence (cDNA). Reverse transcriptase transcribes cDNA into a second strand generating dsDNA. 3. Integration and Transcription : Integrase enzyme catalyses the viral cDNA into the chromosomal DNA of the infected host cell. The integrated DNA is called a 1 Tawit Suriyo provirus. Unincorporated DNA is unstable and decays after a few days. When the host cell is activated, transcription of the viral DNA begins, resulting in the production of multiple copies of viral RNA. These RNA codes for the production of the viral protein a n d e n z y m e s i n t h e t r a n s l a t i o n s t e p . 4. Translation viral RNA to proteins : There are only 9 genes in the HIV RNA. Those genes have the code necessary to produce structural proteins and core enzymes like RT, integrase and a crucial enzyme called a protease. Regulatory genes are expressed early whereas structural genes and genes encoding enzymes are expressed late. 5.Viral protease : Viral RNA is translated into a polypeptide sequence, that includes several individual proteins (RT, integrase and protease). Before these enzymes become functional, they must be cut from the longer polypeptide chain. Viral protease enzyme cuts the long chain into its individual enzyme components which then facilitate t h e p r o d u c t i o n o f n e w v i r u s e s . 6. Assembly and Budding : Finally, viral RNA and associated proteins are packaged and released from the l ymphocyte surface to infect other cells. It is probable that any step in the replication of HIV that is different from cellular replicative process can serve as target for anti-retrovirus therapy, and these includereverse transcriptase, integrase and protease (Figure 1). Reverse transcriptase and protease, inhibitors have been proven to be effective in the clinic, we already have commercial d r u gs o n t h e m a r k e t . H o w e v e r , i n t e g r a s e a s a v i a b l e t a r g e t i s u n d e r . The type of Ani-HIV drug that we have now. S i n c e t h e di s c ov e r y o f H IV a s causative pathogeni c agent of AIDS, a member of potentially useful strategies for the activiral therapy of AIDS and related disease have emerged. In theory, antiviral drugs exert their effects by interacting with viral structural components, virally encoded enzymes, viral genomes or proteins, cellular receptors, cellular enzymes or factor required for viral replication. A growing number of highresolution crystal structures of several HIV-1 enzymes, such as reverse transcriptase, protease and integrase has generated an invaluable database that will be ultimately essential to rationally design antiviral agents. (2,3) Figure 1 : The life cycle of HIV and which step that can stopped. Today we have three classes of anti-HIV drugs : 1. Reverse Transcriptase Inhibitors 2. Integrase Inhibitors ( still in clinical trials) 3. Protease Inhibitors 2 Tawit Suriyo R e v e r s e T r a n s c r i p t a s e I n h i b i t o r s It is the first major class of drugs that is found useful in slowing HIV infection. These drugs can be devided into 2 groups that are Nucleoside analog reverse transcriptase inhibitors (also known as nucleosides, Nukes, or NRTIs) such as Zidovudine (AZT), Didanosine (ddI), Zalcitabine (ddC), Stavudine (d4T), Lamivudine (3TC). Non-nucleoside analog reverse transcriptase inhibitors (also known as nonnucleosides, non-Nukes, or NNRTIs) such as Nevirapine, Delavirdine, E f a v i r e n z . The nucleosides and non-nucleosides both have the same target, i.e. they inhibit (slow down) the action of RT. A RT is important because it changes HIV in a way that lets it become part of the infected cell inside the cell’s command center, its nucleus. If RT doesn’t do its job properly, HIV can’t take over the infected cell and can’t start making new copies of itself. The nucleoside RT inhibitors are all in one group because they work in similar ways. In contrast, non-nucleoside RT inhibitors are completely different from nucl eosi des and t hey i nhi bi t t he act i on of R T i n a di fferent wa ys. ( 1 , 4 ) Nucleoside analog RT inhibitors : The broad family of nucleoside analog RT inhibitors includes 2’,3’ -dideoxynucleosides such as AZT (3’ -azido-2’,3’dideoxythymidine, ddI (2’,3’-dideoxyinosine), d4T (2’,3’ -didehydro-2’,3’dideoxythymidine and ddC (2’,3’-dideoxycytidine). The NRTIs inhibit HIV replication by competitive inhibiton of the incorporation of the natural dNTP substrates of the enzymatic reaction. Second, and more importantly by there own incorporation into the growing viral DNA chain. Incorporation of NRTIs into the viral DNA necessarily leads to DNA chain termination because the NRTIs lack the 3’-hydroxyl group (-OH) in the sugar that is required for further DNA chain elongation. These antiretroviral agents share a common antiviral mechanism of action : they inhibit the RNA dependent DNA polymerase (RT) of human HIV. These agents are both inhibitors and substrates of RT. Despite their common mechanism of action, antiretroviral agents differ substantially in their pharmacologic properties such as rate of absorbtion,distribution and excretion. Including intracellular activation pathways and toxicologic profiles such as differ in intracellular enzyme to metabolite the drugs into active form. Available since 1987, Zidovudine was the first agent shown to provide clinically important benefits. (2,3) Z i d o v u d i n e Zidovudineor AZT is a pyrimidine nucleoside analogue. It is a thymidine analog with antiviral activity against HIV-1,HIV2,human T lymphotropic (leukemia) virus (HTLV)-I and other retroviruses. The chemical name of Zidovudine is 3’-azido-3’deoxythymidine. Zidovudine is a white to beige, odorless, crystalline solid with a molecular weight of 267.24 and a solubility of 20.1 mg/ml in water at 25 o C. The molecular formula is C 10 H13 N5 O4. (5) The structure of Zidovudine and deoxythymidine nucleoside (dThd) are shown in figure 2. 3 Figure 2 : the structure of Zidovudine and deoxythymidine nucleoside. AZT which the 3’-hydroxy group is replaced by an azido group (-N3). Tawit Suriyo The mechanism of action of Zidovudine : After Zidovudine diffuses into host cells, Zidovudine permeates the cell membrane mainly by nonfacilitated diffusion and not via a nucleoside transporter. The drug is initially phosphorylated by cellular thymidine kinase (dTK). Cellular thymidine kinase converts Zidovudine into Zidovudine monophosphate. The monophosphate is further converted into the diphosphate by cellular thymidylate kinase (dTMP-K) and to the triphosphate derivative by other cellular enzymes such as cellular nucleoside 5’-diphosphatekinase (NDP-K). Figure 3. The rate-limiting step is conversion to the diphosphate by thymidylate kinase, so that high levels of the monophosphate but much lower levels of diphosphates and triphosphates are present in cells. The cellular thymidine kinase (EC 2.7.1.21) purified from human lymphocyte H9 cells catalyzes the phosphorylation of dThd and Zidovudine with apparent Km values of 2.9 and 3.0 M, respectively. This means that both compounds have comparable substrate affinities for dTK. The maximal rate of phosphorylation of Zidovudine is equal to 60% of the rate of dThd. For the cellular thymidylate kinase (EC 2.7.4.9), the apparent Km value for AZT-MP is two-fold greater than the Km for dTMP (8.6 M vs 4.1 M), but the maximal phosphorylation rate is only 0.3% of the dTMP rate. AZT-MP should therefore be considered as a substrate inhibitor of dTMP-K, and this conclusion is compatible with the observation that human T lymphocytes incubated with AZT markedly accumulate AZT-MP, whereas only low levels of the 5’-di and 5’-triphosphates are achieved. Incubation of H9 cells with 50 M AZT resulted in intracellular pool levels of AZT-MP as great as 790 M, whereas AZT-DP and AZT-TP pool concentrations were only 4.2 and 1.8 M, respectively. Thus, the intracellular pool levels achieved by AZT-MP in H9 cells are much higher than those of AZT-DP and AZT-TP. Zidovudine triphosphate, which has an intracellular half life of elimination of 3-4 hours, competitively inhibits reverse transcriptase with respect to thymidine triphosphate (TTP). In vitro, Zidovudine triphosphate has been shown to be incorporated into growing chains of DNA by viral RT. Whith Figure 3 : The metabolism of Zidovudine poly(A).oligo(dT)12-18 as the template/primer, Km for dTTP and Ki for AZT-TP values are 2.8 and 0.04 M, respectively. With activated calf thymus DNA as the template, these values are 1.2 and 0.3 M, respectively. When incorporation by the v i r a l e nz ym e o c c u r s , t h e D N A c h a i n i s t e r m i n a t e d . ( 2 , 3 , 5 , 6 , 7 ) F i gu r e 4 . Studies in the cell culture suggest that Zidvudine incorporation by cellular alphaDNA polymerase may occur, but only to a very small extent and not in all test systems. Cellular alpha-DNA polymerase shows some sensitivity to inhibition by the Zidovudine triphisphate with 50% inhibitory concentration (IC50) values 400 to 900 times greater than for HIV-RT. The antiviral selectivity of Zidovudine is due to its greater affinity for HIV reverse transcriptase that human DNA polymerases, althrough low concentrations inhibit DNA polymerase gamma. For DNA polymerase Km for dTTP and Ki for AZTTP are as high as 2.4 and 230 M, respectively, and for DNA polymerase they are 6.0 and 73 M, respectively. Also, to DNA polymerase AZT-TP is much less inhibitory than the closely related 2’,3’-dideoxynucleotide ddTTP (IC50, 11 and 0.1 M, ( 2 ) r e s p e c t i v e l y ) . 4 Tawit Suriyo Several laboratories have reported on the effect of AZT on intracellular deoxynucleotide (dNTP) pools. The physiological relevance of the effects of Zidovudine on dNTP pools with regard to its cellular toxicity are so far unclear. It has been suggested initially that bone marrow toxicity may be due to perturbation of dTTP pools by Zidovudine. This effect may contribute to its cytotoxicity and enhance antiviral effects by decreasing competition for Zidovudine triphosphate incorporation i n t o v i r a l D N A . ( 2 , 3 ) Figure 4 : These are Growing DNA chain and Terminated DNA chain The effective of Zidovudine : Low concentrations ( <0.001 to 0.04 ug/ml) inhibit acute HIV-1 infection in human T-cell lines and peripheral blood lymphocytes. Zidovudine is less active in human monocytes or macrophages but inhibits HIV replication in human brain macrophages and low concentration of Zidovudine inhibit human myeloid and erythroid progenitor cell growth (0.3 to 0.6 ug/ml) and blastogenesis in peripheral blood mononuclear cells. (2) Zidovudine currently is the agent of choice for treatment of HIV infection in patients with CD4 counts less than 500/mm3. Zidovudine is associated with prolongation of survival, decreased opportunistic infections, weight gain, improved functional status, increased CD4 counts and other immunological measures, and decreased HIV plasma RNA. Zidovudine treatment may be benefitial to HIVassociated neurologic disease, thrombocytopenia, psoriasis and lymphocytic interstitial pneumonia. It is uncertian whether lower doses are effective in central nervous system because it can cross the blood brain barrier. Zidovudine is better than most other drugs in attacking the virus in the CNS. (2,3) The frequency and degree of Zidovudine resistance in HIV isolates correlate with stage of infection, CD4 count and duration of therapy. About one-third of isolates from AIDS patients show high-level resistance by 1 year. Patients often harbor mixtures of wild type and resistant variants, and resistant virus may be detected in plasma or peripheral blood mononuclear cells (PBMN) of patients. Resistance is associated with point mutations leading to amino acid substitutions at multiple sites in reverse transcriptase, particularly codons 41,67,70,215 and 219. In vitro resistance to Zidovudine is due to the accumulation on specific mutations in the HIV RT coding region. Five amino acid substitution (Met41Leu, Arg67Asn, Lys70Arg, Thr215Try or Phe, and Lys219Gin) have been described in viruses with decreased in vitro susceptibility to zidovudine inhibition. The extent of resistance appears to be correlated with the ( 2 , 5 ) n u m b e r o f m u t a t i o n s i n R T . 5 Tawit Suriyo Non-nucleoside analog RT inhibitors : The NNRTIs are a structurally heterogenous class of drugs that selectively inhibit HIV-1 replication, often at nanomolar concentration. These agents usually active against AZT-resistant HIV-1 variants but lack activity against HIV-2. The NNRTIs consist of a variety of structurally different classes of compounds that do not need to be metabolized to exert their inhibitory action against HIV. The NNRTIs interact with a nonsubstrate binding site of the HIV-1 RT and conformationally fit into a lipophilic pocket of the enzymes that is 10-15 A distant from the substrate-active site represented by the Asp110, Asp185 and Asp186 triad. As a consequence the NNRTIs are noncompetitive inhibitors of the HIV-1 RT and their inhibitory effect is independent from the endogenous substrate. (3,6) The example drug of this type is Nevirapine (Viramune). Nevirapine is structurally a member of the dipyridodiazepinone chemical class of compounds. The chemical name of Nevirapine is 11-cyclopropyl-5,11-dihydro-4-methyl-6H-dipyrido [3,2b:2’,3’-][1,4] diazepin 6-one. Nevirapine is a white to off-white crystalline powder with molecular weight of 266.3. Molecular form of Nevirapine is C15H14N4O, and the structure o f N e v i r a p i n e i s s h o w n i n f i g u r e 5 . The mechanism of action of Nevirapine : Nevirapine binds directly to RT and blocks the RNA-dependent and DNA-dependent DNA polymerase activities by causing a disruption of the enzyme’s catalytic site. The activity of Nevirapine doesn’t compete with template or nucleoside triphosphate or HIV -2 RT and eukaryote DNA polymerase such as human DNA polymerases alpha,beta,delta,gamma are not inhibited by Nevirapine. (8) Figure 5 : Structure of Nevirapine Integrase Inhibitors Integration is a critical step in the completion of the viral life cycle and subsequent production of new viral particle. Therefore blocking HIV integrase is a viable therapeutic strategy that can abort completion of the viral life cycle, and prevent infection of new, uninfected target cell. To develop an inhibitor, we need to understand the structure of the integrase enzyme (IN) and develop integrase inhibitors that have properties that will specifically inhibit this enzyme. The development of integrase inhibitors is underway with, of course, a long way to go. But the hope is for eventually having an antiviral that works at a point different from both RT and protease inhibitors. (9) The overall integration reaction have 3 steps that are The formation of the initial state complex step (ISC) that is the process of formation of ISC of integrase with viral cDNA. The 3’-processing in reaction step that is the process of removing 2 nucleotides (cytosine and adenine) from the 3’-end to produce new 3’h y d r o x y l e n d s ( C A - 3 ’ O H ) . The DNA strand transfer step that is the process of viral DNA going to host nucleus and joining to host target DNA. 6 Tawit Suriyo HIV-1 integrase (IN) enzyme is a protein of 32 kDa encoded at the 3’-end of the pol gene. IN recognizes specific sequences in the long terminal repeat (LTR) elements of the viral cDNA. The terminal 15 bp of the LTR are necessary and sufficient for sitespecific cleavage and integration. A highly conserved dinucleotide CA repeat immediately upstream of the cleavage site is critical for enzymatic activity. In the first step of the integration reaction, termed 3’-end processing, two nucleotides are removed from each 3’-end to produce new 3’hydroxyl ends (CA-3’OH). This reaction occurs in the cytoplasm, within a large viral nucleoprotein complex. After entering the nucleus, the processed viral dsDNA is joined to host target DNA. HIV integrase is composed of three functional domains. The amino-terminal region (amino acids residues 1-50) whose exact function remains unknown. The central region (amino acids residues 60-160) encodes the catalytic domain for both 3’-processing and DNA strand-transfer activities. The central core domain alone can carry out an apparent reversal of the DNA strand-transfer reaction in vitro, the so-called disintegration reaction. However, both the amino and carboxyl terminal domains are necessary for catalysis of 3’-processing and strand transfer. A single amino acid substitution (F185K) within the catalytic core domain generates a soluble protein that has enabled the core domain to be crystallized and the structure to be solved. The carboxyl-terminal domain (amino acids residues 200-270) is involved in nonspecific DNA binding. Complementation studies with integrase proteins mutated in different domains suggest that the active form of integrase is an oligomer, although the exact s t o i c h i o m e t r y r e m a i n s u n k n o w n . ( 1 0 , 1 1 ) The different approaches to inhibiting the integration process that have been r e p o r t e d a r e 1. Triple helix-mediated inhibition. The IN-binding site located in the U3 LTR ( a sequence unique to the 3’-end of viral)contains a purine motif, 5’-GGAAGGG-3’, that can be selectively targeted by oligonucleotide/intercalator conjugates (oligopurinesoxazolopyridocarbazole). Under neutral pH and at physiological temperature, these conjugates readily form a stable complex with the viral DNA that involves a short DNA triplex. It has been shown that triple-helix formation at this site can prevent the catalytic f u n c t i o n o f I N i n v i t r o . 2. Inhibition by peptides derived from combinatorial peptide libraries. That is the process of changing amino acids in the critical portion of the IN enzymes and make the I N l o s s o r d e c r e a s e i t s f u n c t i o n . 3. Screening of chemical libraries and natural compounds. A number of inhibitors that show activity in this sort of oligonucleotide-based in vitro assays have been reported They belong to three main categories: DNA-binding agents, polyhydroxylated aromatic compounds, and nucleotides. Many DNA-binding agents were found to inhibit HIV-1 IN, probably due to a nonspecific interaction with the DNA-binding domain of the enzyme. Polyhydroxylated aromatic compounds are believed to interact with the catalytic domain, possibly by interfering with the coordination of the metal ions required for the phosphoryl transfer reaction. And nucleotides such as 3’-azido-2’,3’-dideoxy-TMP interfere with the enzymatic activity of IN, possibly by binding to the polynucleotide binding site. 4. Inhibition by G-quartet forming oligonucleotides. Recently, the classes of oligonucleotides composed of deoxyguanosine and thymidine were reported to inhibit HIV-1 replication in cell culture. The first class of oligonucleotides consists of 16- or 17mers that contain single phosphorothioate internucleoside linkages at their 5’- and 3’ends. In the presence of potassium, these oligonucleotides can fold upon themselves to form a highly stable four stranded DNA structure containing two stacked G-quartets. Two possible mechanisms for the observed antiviral activity have been proposed: inhibition of viral entry into the cell and/or inhibition of HIV -1 IN. The phosphorothioate 7 Tawit Suriyo oligonucleotide TTGGGGTT, which adopts a parallel-stranded tetrameric G-quartet structure, is also a potent inhibitor of HIV replication in cell culture. This compound exerts its antiviral effect through potent and specific inhibition of HIV envelope-mediated ( 1 0 ) v i r u s b i n d i n g a n d c e l l f u s i o n . One integrase inhibitor that has make it into clinical trials is AR-177 made by Aronex. The brand name is Zintevir (T30177 or AR177). Zintevir is a stable oligonucleotide of 17 nucleotides in length. It is a potent inhibitor of HIV and inhibit the action of the integrase enzyme. The molecular weight of AR177 is 5,793 daltons. Zintevir is composed only of deoxyguanosine and thymidine, with single phosphorothioate internucleoside linkages at its 5’ and 3’ ends. The sequence of Zintevir is 5 ’ - G T G G T G G G T G G G T G G G T - 3 ’ The mechanism of action of Zintevir : Two possible mechanisms of Zintevir for the observed antiviral activity have been proposed : interference with virus adsorption to the cell and/or inhibition of HIV-1 integrase. The sequence of Zintevir which impart and compact tetrad structure believed to be responsible for its potent inhibition of HIV-1 integrase activity. Zintevir that can form a highly stable intramolecular four-stranded DNA structure containing two stacked guanosine-quartets (G-quartets) has been reported to inhibit the replication of the HIV-1 in cell culture. Figure 6. The mode of action of this compound, which forms a guanosine-quartet structure has been attributed to G-quartet structures that may act as negative regulators of autointegration in vivo. Novel AIDS therapies could be based upon G quartets as inhibitors of HIV integrase. The molecular interaction of Gquartet-containing oligonucleotides with Figure 6: Sequence and structure of G- HIV-1 integrase. The prototypical Gquart et form ing ol ignucl eoti des of quartet cotaining oligonucleotide, Z i n t e v i r , t h a t c o n t a i n s i n g l e T30177 (zintevir), inhibited the phosphorothioate internucleoside linkage integration reaction by, Zintevir inhibits at the 3’- and 5’- ends. and folding pattern the step of the formation of the ISC, for a Zintevir monomer with the formation while it does not inhibit 3’-processing or o f t w o G - q u a r t e t s . DNA strand transfer. Zintevir was capable of preventing the binding of integrase to specific DNA by the interaction of HIV-1 IN with G-quartets resulting in the inhibition of the formation of the ISC between IN and substrate DNA. Zintevir is merely competing with the specific DNA substrate for binding to IN. The inhibition by Zintevir was not affected by the increase in substrate concentration. It has been suggested, that Zintevir has direct interactions with the enzyme IN not with the substrate DNA. It is possible that the G-quartets interact with IN before the enzyme forms oligomers capable of a functional interaction with the LTR (long terminal repeated sequence) DNA. Alternatively, Zintevir may block the binding of IN oligomers to the LTR. The oligomerization of IN, interaction with the LTR, and maturation of the complex are likely to be interconnected processes. Because the inhibition by G-quartets cannot be competed out by the addition of an excess LTR, it is unlikely that the compounds bind to the DNA binding domain. Interestingly, a recent report suggests an interaction of Zintevir with the zinc finger domain of IN. this domain 8 Tawit Suriyo has been invoked in the functional oligomerization of HIV-1 IN. Alternatively, these Gquartet-containing oligonucleotides may interfere with entry of the virus in the target cell. ( 1 0 , 1 2 , 1 3 ) Protease Inhibitors Protease inhibitors get their name because they slow down the action of another HIV enzyme, protease. Protease goes to work inside infected cells after proteins made by HIV come out of the nucleus. It works like a "chemical scissor" cutting up these long chains of HIV proteins and enzymes (polypeptide) into smaller pieces (functional enzyme). HIV needs these smaller pieces to make active new copies of itself. Protease inhibitors gum up the protease "scissors." The result is that new copies of HIV aren't made the right way (the new copies of HIV are empty not have smaller protein and enzyme inside the cell) and they can't go on to infect new cells. Examples of drugs that are in the m arket are Rit onavi r, Indi navi r, S aquinavi r, Nelfinavir. ( 1 , 4 ) Protease enzymes are proteins that cut other proteins at highly specific locations. The HIV protease is an aspartyl protease enzyme similar to mammalian proteases like renin. HIV-1 aspartate protease in its active form consists two symmetric subunit (identical subunit), and consists of 99 amino acids that join symmetrically to form the functional enzyme. The funtional protease exists as a C2-symmetric homodimer with a single active site. Each monomeric unit contributes one of the conserved catalytic triads (Asp-Thr-Gly) common to the aspartic protease class. This enzyme is required for cleavage of polypeptide precursors that generate the structural proteins and enzymes of the virus, including RT, integrase and the protease itself. The HIV protease enzyme cleaves polyproteins of the virus into essential functional protein products during the maturation process of the virion. If a mutation occurs in the catalytic aspartate residues to either aspargine or alanine, the generated viral particle with remained in the immature form. This critical process occurs as each new virion buds forth from the membrane of an HIVinfected cell and continues after the immature virus is released from the cell. If the polyproteins are not cleaved, the virus fails to mature and is incapable of infecting a new ( 3 , 1 1 , 1 4 , 1 5 ) c e l l . A number of protease inhibitors, based on transition state mimetics of peptide substrates have been described. Other inhibitors interact with catalytic residues and displace a structural water molecule. The protease inhibitors represent one of the best examples of the application of basic scientific knowledge to rational drug design. Relatively soon after the discovery of HIV, researchers were able to elucidate the genetic structure of the virus (a process that identified an array of structural and regulatory gene products). Within the pol region of the virion's genetic structure, researchers found codons for HIV protease, reverse transcriptase, and integrase, which are essential to the maturation of the virus. Although the genetic sequence of the HIV protease enzyme was described soon after the discovery of the virus itself, the three-dimensional structure of the gene was not demonstrated until several years later, when the protease gene product was expressed and crystallized. However, researchers were able to begin designing compounds that specifically targeted the active site of the HIV protease. HIV protease activity is unique for HIV , and in the host there is virtually no cross-reactivity between the HIV protease and normal human protease gene products. It is this lack of cross-reactivity that gives the protease ( 1 6 , 1 7 ) inhibitors their outstanding safety profile. 9 Tawit Suriyo The protease inhibitors are able to inhibit the function of the native protease enzyme. They exert their inhibitory effect by disabling the enzyme before it can cleave the gag/pol polyprotein into its essential products. Like a key perfectly fitted to a lock, the protease inhibitor simply locks up the enzyme, rendering it ineffective. The inhibitors will interact directly with the active site of protease. Figure 7. This interaction between protease and protease inhibitor causes the enzyme protease to not function properly. Native protease Inhibitor interact with HIV protease Figure 7 : Native HIV protease and protease with inhibitor The majority of HIV protease inhibitor classes are peptidomimetics, refleting the structure of the polyprotein substrates of HIV protease. For example Ritonavir, Saquinavir, Indinarvir, and Nelfinavir. HIV Ritonavir (Norvir,ABT-538) a symmetry-based HIV protease inhibitor with a substantially reduced rate of metabolism and high oral bioavilablility in animals and humans. Ritonavir is chemically designated as 10-Hydroxy-2-methyl-5-(1-methylethyl)1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12tetraazatridecan-13-oic acid,5-thiazolylmethyl ester, [5S-(5R*,8R*,10R*,11R*)]. Its molecular formular is C37H48N6O5S2 and its molecular weight is 720.95. Ritonavir is a white-to-light-tan powder. Ritonavir has a bitter metallic tase. It is freely soluble in methanol and ethanol, soluble in isopropanol and practically insoluble in water. (18) The s t r u c t u r e o f R i t o n a v i r i s s h o w n i n f i g u r e 8 . Ritonavir is a peptidomimetic inhibitor of both the HIV-1 and HIV2 protease. Inhibition of HIV protease renders the enzyme incapable of processing the gag-pol polyprotein precursor which leads to production of noninfectious immature HIV particles. Ritonavir produces dramatic declines in plasma HIV Figure 8 : The structural formula of Ritonavir RNA and substantial recovery in CD4 cell levels in HIV-infected individuals Accompanying these changes in surrogate markers, administration of Ritonavir is associated with longer life-expectancy The mechanism of action of Ritonavir : The antiviral activity of Ritonavir by the additional hydrophobic interaction of the P3 isopropylthiazolyl group of Ritonavir with active side chain of valine-82 (V82) of HIV protease. The preliminary X-ray crystal structure of Ritonavir bound to HIV protease confirms this interaction and rationalizes the structure-activity relationships. The close interaction of the P3 isopropyl group with V82 also appears to affect the response of HIV to selective pressure during treatment with Ritonavir. Figure 9. HIV strains mutated at valine-82 to alanine, phenylalanine, and threonine have been shown to be the primary species present during the initial rebound of plasma HIV RNA during suboptimal monotherapy with Ritonavir. 10 Tawit Suriyo Ritonavir is principally metabolized by the liver, by enzyme cytochrome P450 (CYP 3A4) and its also a potent inhibitor of the CYP 3A4. Inhibition of CYP by Ritonavir is associated with the unhindered nitrogen atom on the unsubatituted P2’ 5’-thiazolyl group, which binds directly to the heme in the CYP a c t i v e s i t e . ( 1 6 , 1 7 , 1 8 ) Figure 9 : This is the interaction between side chain of valine-82 of HIV protease with Ritonavir C o n c l u s i o n Human Immunodeficiency Virus depends on the cells that it infects to make new copies of itself. These new copies of HIV go on to infect other cells. Now the step that we can stop HIV replication are reverse transcriptase, integrase and protease. Two of them Reverse transcriptase inhibitors (RTIs) and protease inhibitors(PIs), already have commercial drugs on the market. But for integrase inhibitors(INs), its still under go in the clinical trial. RTIs, it can be devided into 2 groups that are Nucleoside analog (e.g. AZT,ddI.ddC) and non-nucleoside analog ( e.g. Nevirapine, Delavirdine). Both of them have the same target, they inhibit the action of the HIV RT enzymes. But they inhibit the action of RT in a different ways. NRTIs competitive inhibition of the incorporation of the natural dNTP substrates. And its incorporation into the growing viral DNA chain, leads to DNA chain termination. These agent are both inhibitors and substrates of RT, and they need metabolism to exert their inhibitory function. For the NNRTIs, they are a structurally heterogenous class of drugs that selectively inhibit HIV-1 replication only. They do not need to be metabolized to exert their inhibitory effect. The NNRTIs interact with a non-substrate binding site of RT, as a consequence they are non-competitive inhibitor and their inhibitory effect is independent from the endogenous substrate. For INs, the different approaches to inhibiting the integration process that have been reported are triple helix-mediated inhibition (this formation can prevent the catalytic domain of IN), inhibition by peptides derived from combinatorial peptide libaries, sreening of chemical libraries and natural compounds that inhibit function of IN, and inhibition by Gquartet forming oligonucleotides. For PIs (e.g. Ritonavir, indinarvir,nelfinavir), they are able to inhibit the function of the native HIV protease enzymes. The PIs simply locks up the enzyme, rendering it ineffective. The inhibitors will interact directly with the active site of protease. This interaction between protease and protease inhibitor causes the e n z ym e s p r o t e a s e t o n o t f u n c t i o n p r o p e r l y. 11 Tawit Suriyo R e f e r e n c e s 1. Martin Markowitz. Combination Therapy for HIV Infection. International Association P h y s i c i a n s i n A I D S C a r e ( I A P A C ) . 1 9 9 7 . 2. Samnel Broder, Thomas C. Merigan, and Dani Bolognesi. Text Book of AIDS M e d i c i n e . W i l l i a m s & w i l k i n s . 1 9 9 4 . 3. Alfred Goodman Gilman. GOODMAN & GILMAN’s the Pharmacological basis of therapeutics. 9th edition. McGraw-Hill. 1996. 4. H I V - A I D S i n f o r m a t i o n b y A E G I S . h t t p : / / w w w . a e g i s . c o m 5. R E T R O V I R P a t i e n t I n f o r m a t i o n . 6. Jan Balzarini, Lieve Naesens and Erik De Clercq. New antivirals-mechanism of a ct i o n an d r esi st an c e d e ve l op m e nt . Mi cro bi o l og y , 1: 535 -5 4 6 (19 98 ). 7. Jan Balzarini, Lieve Naesens, S. Aaquaro, T. Knispel, C.-F. Perno, E. De Clercq, and C. Mei er. Int racel lular Metabol ism of C ycloS ali gen yl 3’ -Azido -2’,3’dideox yth ymidineMonophosphate,a Prodrug of 3’ -Azido-2’,3’d i d e o x y t h y m i d i n e ( Z i d o v u d i n e ) . 1 9 9 9 . 8. V I R A M U N E P a t i e n t I n f o r m a t i o n . 9. Mark Katz . Medical update: Int egrase Inhibitors. Bei ng Ali ve. 1995. 10. Peter Cherepanov, Jose A. Este, Robert F. Rando, Joshua O. Ojwang, Gunther Reekmans, Robert Steinfeld, Guldo David, Erik De Clercq, and Zeger Debyser. Mode of Interaction of G-Quartets with the Integrase of Human Immunodeficiency Virus Type1. Molecular Pharmacology,52:771:780(1997). 11. Bernard N. Fields, David M. Knipe, and Peter M. Howlen.Fundamental Virology. 3rd e d i t i o n . L i p p i n c o l l - R a v e n . 1 9 9 6 . 12. Jose A. Este, Cecilia Cabrera, Dominique Schols, Peter Cherepanov, Arantxa Gutierrez, Myriam Witvrouw, Christophe Pannecouque, Zeger Debyser, Robert F. Rando, Bonaventura Clotet, Jan Desmyter, and Erik De Clercq. Human Immunudeficiency Virus Glycoprotein gp120 as the Primary Target for the Antiviral Action of AR177(Zintevir). Molecular Pharmacology,53:340-345(1998). 13. Thomas L. Wallace, Christina Gamba-Vitalo, Ken S. Loveday, and Paul A. Cossum. Acute, Multiple-Dose, and Genetic Toxicology of AR177, and Anti -HIV Oligonucleotide. Toxicological Science, 53:63 -70 (2000). 14. Martin Markowitz. Protease Inhibitors. . International Association Physicians in AIDS C a r e ( I A P A C ) . 1 9 9 7 . 15. Dale J. Kempf, Hing L. Sham, Kennan C. Marsh, Charles A. Flentge, David Betebenner, Brain E. Green, Edith McDonald, Sudthida Vasavanonda, Ayda Saldivar, Norman E. Wideburg, Warren M. Kati, Lisa Ruiz, Chen Zhao, LynnMarie Fino, Jean Patterson, Akhteruzzaman Molla, Jacob J. Plattner, and Daniel W. Norbeck. Discovery of Ritonavir, a Potent Inhibitor of HIV Protease with High Oral Bioavailability and Clinical Efficacy. Journal of Medicinal Chemistry,41:602-617 ( 1 9 9 8 ) . 16. Amy S. Ripka, Daniel H. Rich. HIV protease. Chemical Biology,2:441-452 (1998). 17. How the Protease Inhibitors Work. http://www.thebody.com/hivnews/newsline/june96/pullout.html 18. Norvir Patient Information. 12