A Tutorial of “Cross-Phenotype Normalization by Iterative Nonlinear
advertisement

A Tutorial on Cross-Phenotype Normalization by Iterative Nonlinear Regression
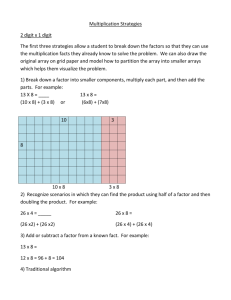
1. Introduction
baseline array
baseline array
baseline array
baseline array
The cross-phenotype normalization (CPN) method by iterative nonlinear regression (INR) has been
implemented in C/C++ and integrated into DNA-Chip Analyzer (dChip) software (Li and Wong, 2003) for
normalization of microarray data. Fig. T-1.
shows a snapshot of the CPN-INR method and
dChip software working together for microarray
data analysis. dChip is a software package that
implements model-based expression analysis of
oligonucleotide arrays (Li and Wong, 2001). It
also implements many low-level data analysis
routines (e.g., data I/O, gene filtering, etc.) and
several high-level analysis procedures (e.g.,
clustering and classification). Normalization by
invariant ranking (IR) was also implemented as
one of the low-level routines, which is of
particular interest for us to compare our CPNINR method (Xuan et al., 2005) to IR (Schadt et
al., 2001). The dChip program is a single
executable program running on Windows, under
which it was originally developed in Wong’s
Lab at Harvard University.
Fig. T-1. A snapshot of the INR method and dChip for data
The novel feature of our CPN-INR
analysis.
method is that the method unifies the tasks of
estimating normalization regression function and
identifying invariantly expressed genes (IEGs)
for normalization. An iterative IEG selection
algorithm was developed to couple two processes
so that the regression function estimation and
IEG selection bootstrap one another (Wang et
al., 2002, Xuan et al., 2005). Fig. T-2 shows an
example of the iterative IEG selection process in
floating array
floating array
action for normalization.
In this tutorial, we will briefly describe
the CPN-INR algorithm and use an example to
illustrate the usage of this utility. In particular,
Section 2 will summarize the block diagram of
the CPN-INR algorithm. In Section 3, we will
describe the procedure of using INR for
normalization of a microarray data set. It is
strongly suggested that the users shall follow the
floating array
floating array
steps described in the procedure to explore the
Fig.
T-2.
Iterative
IEG
selection
selected
IEGs
are in red.
functionalities of CPN-INR.
2.
A Brief Description of the CPN-INR Method
As shown in Fig. T-3, the CPN-INR
algorithm has been implemented as a two-step
Phenotype A (Floating)
Phenotype B (Baseline)
boosting approach by iteratively performing
Within-Phenotype
Within-Phenotype
the
followings:
(1)
within-phenotype
Normalization
Normalization
normalization and (2) between-phenotype
Sector-Shaped IEG Selection
Sector-Shaped IEG Selection
normalization. In the first step (withinNormalization by Nonlinear
phenotype normalization), INR method is
Normalization by Nonlinear
Regression Based on
Regression Based on
Selected IEGs
applied to all the arrays within a phenotype for
Selected IEGs
normalization. In the second step (betweenNo
No
Converged?
Converged?
phenotype normalization), a within-phenotype
Yes
Yes
Normalization by Nonlinear
Normalization by Nonlinear
“mean profile” is first generated from the
Regression
Based
on
Regression Based on
Final Selected IEGs
Final Selected IEGs
normalized arrays for each phenotype. The
CPN-INR method is then applied to these
Generate Within-Phenotype
Generate Within-Phenotype
within-phenotype “mean profiles” to select the
“Mean” Profile
“Mean” Profile
IEGs and estimate the regression function. The
Cross-Phenotype
estimated regression function will then be
Normalization
Sector-Shaped IEG Selection
applied to all the arrays in “floating”
phenotype for normalization. This process
Normalization by Nonlinear
Regression Based on
cycles back and froth until the following two
Selected IEGs
No
conditions are met: (1) the selected ISGs differ
Converged?
from previously selected ISGs very little, and
Yes
Estimate Nonlinear
(2) the estimated regression function
Regression Function Based on
Final Selected IEGs
approaches to a 45-degree line shown in scatter
Apply Estimated Regression Function
plot.
to All the Arrays in “Floating” Phenotype
In practice, CPN-INR method has been
No
implemented in a way that normalization can
Converged?
be carried out either at probe level for all the
Yes
probes on oligonucleotide array data, or at
gene level for all the genes on cDNA array.
Fig. T-3. Block diagram of the CPN-INR algorithm.
When carried out in probe level, we will only
use perfect match (PM) probes to select IEGs (or to be exact, invariantly expressed probes (IEPs)). Note that
this is consistent to the implementation of iterative ranking (IR) method (Schadt et al., 2001), but different from
Bolstad’s implementation where both PM and mismatch (MM) probes are used for selecting invariant genes
(Bolstad et al., 2003).
3. A Procedural Description of the CPN-INR Method
In this section, we will describe a procedure to perform normalization of microarray arrays by using
CPN-INR. The following steps are described and illustrated in details: (1) preparing data for normalization, (2)
reading in data, (3) normalizing arrays using CPN-INR, (4) exporting normalized data to files, and (5) plotting
normalization function.
3.1.
Prepare Data
To use dChip, the user needs to provide Affymetrix
array data files (in CEL or DAT format) and the CDF file
(Chip Description File). If it is desired to read in cDNA
array data or gene expression data like those generated by
MAS5.0, one may make an external data using tabdelimited text format, and then read it into dChip by
“Analysis/Get external file”.
To use CPN-INR for normalizing cell files among
multiple phenotypes, it is required to create a directory that
contains the subdirectories for the phenotypes. Each
subdirectory will hold all the cell arrays in one-specific
phenotype. To normalize gene expression data (external
file), we will create separate tab-delimited text file for each
phenotype. Note that an upcoming version is also under
development, which can use label to assign each sample to
a particular class (or phenotype).
3.2.
Read in Data
dChip analysis is based on a group of array data
files a researcher generates, either in DAT or CEL format.
All the arrays to be used in a single analysis should be of
the same chip type. To read in the data, select the menu
“Analysis/Open Group”. With a dialog window shown in
Fig. T-4, the user can give a group name, select a data
directory and specify the data format of the files to be read
in.
After completing the “Analysis/Open group/Data
files” dialog, click “Other information” tab on the top and
another dialog will be shown (Fig. T-5). In this dialog, the
CDF (Chip Description File) file for a particular array type
should be specified. See Affymetrix website (URL:
http://www.affymetrix.com) for obtaining the CDF files.
To read in a group of multiple phenotype arrays,
click “Analysis/Open Supergroup (CPN)” to specify each
group’s name, data directory, file format, and CDF as
shown in Fig. T-4 and T-5. For each group, a dialog
window will be prompted for user to specify the
information and the dChip software will read in the data
when the user clicks “OK”. After all groups of arrays read
in, click “Cancel” to signal the completion of data “read
Fig. T-4. The dialog window of “Analysis/Open
group/Data files”.
Fig. T-5. The dialog window of “Analysis/Open
group/Other information”.
in” process. A similar procedure can be used to read in gene expression data files in a tab-delimited txt format
for CPN normalization.
Click “OK” to perform read-ins and in the output we can read some brief information about (1) the CDF
file and (2) a summary of the arrays indicating median intensities and percentage of “Present Call”:
{Open group GL_SNB30
Read in existing binary CDF file G:\NormMS4Final\CDF\HG_U95A.cdf.bin (file format 3)...
Search and extract PM/MM data from 'CEL' files of chip type 'HG_U95A' in
G:\NormMS4Final\Variance\GL_SNB30\TuT...
Found G:\NormMS4Final\Variance\GL_SNB30\TuT\94424hgu95a11.cel
Reading CEL file...
Line 400000
Calculating presence calls...
Gene 12600: P
Found G:\NormMS4Final\Variance\GL_SNB30\TuT\94425hgu95a11.cel
Reading CEL file...
Line 400000
Calculating presence calls...
Gene 12600: A
Found G:\NormMS4Final\Variance\GL_SNB30\TuT\94426hgu95a11.cel
Reading CEL file...
Line 400000
Calculating presence calls...
Gene 12600: P
Found G:\NormMS4Final\Variance\GL_SNB30\TuT\94427hgu95a11.cel
Reading CEL file...
Line 400000
Calculating presence calls...
Gene 12600: P
Calculating background...
Writing array summary file G:\NormMS4Final\IEGOverlap\GL_SNB30\GL_SNB30 arrays.xls
Group "GL_SNB30" contains 4 files, see G:\NormMS4Final\IEGOverlap\GL_SNB30\GL_SNB30
arrays.xls for details
Number
1
2
3
4
Array
File Name Median Intensity
P call %
94424hgu95a11
G:\NormMS4Final\Variance\GL_SNB30\TuT\94424hgu95a11.cel
235 59.1874
94425hgu95a11
G:\NormMS4Final\Variance\GL_SNB30\TuT\94425hgu95a11.cel
209 58.8389
94426hgu95a11
G:\NormMS4Final\Variance\GL_SNB30\TuT\94426hgu95a11.cel
222
58.633
94427hgu95a11
G:\NormMS4Final\Variance\GL_SNB30\TuT\94427hgu95a11.cel
170 58.7835
Treat 4 arrays as 4 experiments
Finished in 0 minutes 24 seconds}
3.3.
Normalize Arrays
Normalization is an important prerequisite for almost all follow-up microarray data analysis steps. Since scanned
images may have different overall brightness, normalization is needed to adjust the brightness of the arrays to
comparable level. To normalize multiple arrays, click “Analysis/Normalize” to bring up the normalization
dialog as shown in Fig. T-6. As we can see, the CPN-INR method has been implemented as an alternative
method in dChip in addition to IR method. With the dialog, we can select several parameters for INR method
including (1) baseline array and (2) different forms of nonlinear regression function.
Both INR and IR methods are baseline-based
normalization methods. By default an array with median
overall intensity is chosen as the baseline array against
which other arrays are normalized at probe intensity level.
Using the normalization dialog we can specify a different
array as the baseline if necessary. For the different forms
of nonlinear regreesion function, we have implemented the
followings: (1) quadric polynomials, (2) cubic polynomials
and (3) smoothing spline with generalized cross-validation
(GCVSS). Among them, cubic polynomials possess some
advantage over quadratic polynomials and GCVSS due to
its accuracy in model fitting and its low computational
complexity in model parameter estimation.
Notice that normalization in dChip will first check
if arrays were normalized before. If normalized, dChip will
use existing normalized CEL intensities in DCP files. To
force dChip to always do normalization, check “Ignore
normalized data in DCP files”. Click “OK” to perform
Fig. T-6. A snapshot of the INR method within dChip
normalization and in the output we can read how many PM
for data analysis.
probes are selected in the “Invariant set” (i.e., the IEGs) to
determine the normalization curve, and the median probe intensity before and after normalization:
{Normalize arrays using baseline: 94426hgu95a11...
Baseline chip
Accessing 'G:\NormMS4Final\Variance\GL_SNB30\TuT\94426hgu95a11.dcp'
94424hgu95a11
Accessing 'G:\NormMS4Final\Variance\GL_SNB30\TuT\94424hgu95a11.dcp'
Searching Invariant-set: 16290
Normalizing CEL: 400000
Median intensity: 235 -> 201
94425hgu95a11
Accessing 'G:\NormMS4Final\Variance\GL_SNB30\TuT\94425hgu95a11.dcp'
Searching Invariant-set: 18710
Normalizing CEL: 400000
Median intensity: 209 -> 218
94426hgu95a11
Accessing 'G:\NormMS4Final\Variance\GL_SNB30\TuT\94426hgu95a11.dcp'
Do not normalize the baseline array (Median intensity 222)
94427hgu95a11
Accessing 'G:\NormMS4Final\Variance\GL_SNB30\TuT\94427hgu95a11.dcp'
Searching Invariant-set: 19041
Normalizing CEL: 400000
Median intensity: 170 -> 199
Calculating background...
Finished in 0 minutes 58 seconds}
(file format 3)
(file format 3)
(file format 3)
(file format 3)
(file format 3)
3.4.
Save Normalized Data
After normalization, the user may use “Image/Export
CEL” to export probe level data of one or all arrays into
CEL files. The dChip-generated CEL file does not have the
standard deviation and number of pixel information in the
original CEL file. As a result Affymetrix MAS5 software
cannot recognize dChip-generated CEL files. We have
implemented a temporary/alternative way to remedy this
problem by filling in “fake” numbers for the standard
deviation and number of pixel information. Click
“Image/Export CEL(Mas5.0)” to bring out the dialog shown
in Fig. T-7. The user can specify to save either the current
normalized array or all normalized arrays. For the
normalized CEL files, the user can specify a word to be
inserted into the file names.
3.5.
Fig. T-7. Export normalized intensities to CEL files.
Plot Normalization Function
When in the “Image View” (where an array image is displayed), one can use “Image/Normalization plot”
(Installation of R software is needed, see R-Project, (2005) for more information about the R software) to view
the normalization scatter plot between the current array and the baseline array. Fig. T-8 shows the normalization
plot dialog where the user can specify whether to use smoothing spline for computing the normalization curve
or not. Fig. T-9 shows a scatter plot displayed by R software. In the plot, each point represents PM or MM
probe values in the two arrays. The blue line is the diagonal line y = x, the red circles are the probes selected in
the IEG set, and the green curve is the normalization curve based
on the selected IEGs.
Fig. T-8. Normalization scatter plot.
4.
Summary
Fig. T-9. Scatter plot of two arrays before
normalization. The IEGs selected by INR are
shown in red, and the estimated normalization
function is shown in green. The blue line is at
45-degree.
In this tutorial, we have presented the CPN-INR method
with a procedural description of the method. The CPN-INR
method has been implemented in C/C++ and integrated into dChip software. The block diagram of the CPNCPN-INR algorithm has been summarized in Section 2. The procedural steps for performing normalization
using CPN-INR have been detailed and illustrated in Section 3.
References
1.
2.
3.
4.
5.
6.
7.
Li, C., and Wong, W.H. (2003) DNA-Chip analyzer (dChip). In The analysis of gene expression data: methods
and software. Edited by Parmigiani, G., Garrett, E.S., Irizarry, R. and Zeger, S.L. Springer.
Li, C., and Wong, W.H. (2001) Model-based analysis of oligonucleotide arrays: model variation, design issues
and standard error application. Genome Biol., 2(8) 1-11.
Xuan, J., Khan, J., Hoffman, E., Clarke, R., and Wang, J. (2005) Normalization of microarray data by iterative
nonlinear regression. Submitted to IEEE 5th Symposium on Bioinformatics and Bioengineering (BIBE05).
Schadt, E., Li. C., Eliss, B., and Wong, W.H. (2001) Feature extraction and normalization algorithms for highdensity oligonucleotide gene expression array data. J. Cell. Biochem., 84(S37), 120-125.
Wang, Y., Lu, J., Lee, R., Gu, Z., and Clarke, R. (2002) Iterative normalization of cDNA microarray data. IEEE
Trans. on Info. Tech. in Biomedicine, 6(1), 29-37.
Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. (2003) A comparison of normalization methods for
high density oligonucleotide array data based on variance and bias. Bioinformatics, 19(2), 185-193.
The R-Project (2005) The R project for statistical computing, URL: http://www.r-project.org/.