10_11_rel_corr_PbC_2

AB INITIO CALCULATIONS ON THE INTERATOMIC POTENTIAL OF PB – C
DIATOMIC TAKING INTO ACCOUNT RELATIVISTIC EFFECTS
V. Kuzmin
Vast variety of applications ranging from microelectronics to aerospace and automotive industries and also biomedical technologies uses ion beam techniques for modification of different materials. Amongst other carbon and carbon materials take a special place, being of great importance for modern as well as for future technologies and in particularly for nanotechnology. Considerable interest in producing nanoscale objects make increasing demands of accuracy of theoretical predictions. However, considerable differences
(more than 40%) between experimental data
1-
3
and the values given by Ziegler, Biersack and Littmark (ZBL) theory
4
for range parameters of heavy ions in light targets at energies of about 1-1000 keV were revealed in recent years. These discrepancies were explained by a correlation between the nuclear and electronic energy losses
1,2
. In addition to the quasielastic description of atomic scattering in those works it was proposed to transform the universal ZBL potential with the ZBL electronic stopping power. It should be stressed, that the universal ZBL potential is an average of a large number of repulsive potentials, calculated with an approximate method
4
. There-
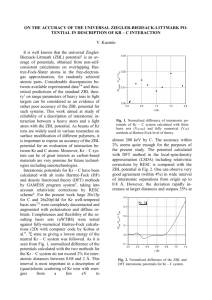
Fig. 1.
Normalized difference of interatomic potentials of Pb – C system calculated with finite basis sets (uWTBS) and fully numerical
(2D) methods at Hartree-Fock level of theory.
Large 20s13p for C and 28s24p18d12f for Pb well-tempered basis sets 7,8 (WTBS), augmented with polarization and diffuse orbitals, have been employed in the present work.
Completeness and flexibility of the basis sets were tested against fully-numerical restricted
Hartree-Fock calculations with computer code by Kobus et al.
9
. Electronic configurations of neutral diatomic with lowest spin multiplicity were considered. As it is seen from Fig. 1, normalized difference of the potentials calculated with the two methods at Hartree-Fock level do not exceed 1.5% in the range of interatomic distances of 0.08 – 1.2 Å. For Pb fore, the large discrepancies between the experimental and theoretical values for range parameters can indicate the lack of accuracy of the ZBL potential and/or the ZBL electronic stopping for ion-target combinations in question.
In the present work interatomic potentials for Pb – C diatomics have been calculated with Hartree-Fock and density functional theory (DFT) methods by GAMESS program system
5
including relativistic corrections by
RESC scheme 6 , and compared with the universal ZBL potential. GAMESS realizes finite basis set approach using Gaussian basis sets.
The package as well as basis sets were developed for problems of quantum chemistry i.e. for a consideration of systems nearby (quasi)equilibrium state. Therefore great care should be taken to select proper basis sets. ions with energies from a few keV to almost 1
MeV bombarding a carbon target these separations are determining in a description of
(quasi)elastic scattering.
It is well known that relativistic effects are important for heavy atoms. Using the uncontracted well-tempered basis sets (uWTBS), screening functions have been calculated with
Hartree-Fock method including relativistic corrections by RESC scheme. Similar calculations with DFT at local spin density approximation (LSDA) level have been performed.
The results of the calculations are shown in
Fig. 2. Also, screening functions for the uni-
Fig. 2.
Screening function for Pb – C diatomic calculated with DFT and HF methods including relativistic corrections by RESC scheme using uncontracted WTBS basis sets (see text). Those for the ZBL and Molière potentials are given for comparison. The inset shows a behavior of the screening functions at larger distances. versal ZBL and the Molière potentials are given for comparison. It is interesting to note that in accordance with Ref. 4 the average of the relative errors squared for the Molière potential is 237% whereas for the ZBL potential that is only 5%. The difference between the
DFT and the ZBL potentials reaches 11% for separations r
≤ 0.529 Å (Bohr radius) and rapidly increases with interatomic distance up to
46% at 1.2 Å. Relativistic corrections yield of
Fig. 3.
The nuclear stopping powers of Pb ions in
C as determined by DFT potential (filled circles), by Molière-like fit to HF potential (dash-dotted line) and that of SRIM2003 (solid line). Also, the electronic stopping powers given by SRIM2003
(dashed line) and by Land and Brennan tables 10
(dots) are plotted for comparison. about of 3% at the former separation and of
13 % at the latter one.
The differences in the potentials lead to notable changes in the nuclear energy loss. In
Fig. 3 the nuclear stopping powers as determined by the DFT, by Molière-like fit to HF and by the ZBL potentials are presented. Also, the electronic stopping powers from
SRIM2003 package and by Land and Brennan
10
are given for comparison. One can see that the nuclear stopping power as determined by the ZBL potential is approximately 10% larger than that of first principles calculation in a wide interval of energies above 200 keV.
At energies below 1 keV the differences exceed 20% increasing up to 40% at 100eV.
However at energies below a few hundreds eV one needs also to take into account manybody character of the interactions.
We conclude that in order to obtain a more accurate description of range and damage distributions induced by a heavy ion implantation in carbon materials one needs to use the correct interatomic potentials, which can be derived from the first principles calculations taking into account relativistic corrections.
REFERENCES
1) M. Behar et al., Nucl. Instrum. Meth.,
B59/60 (1991), 1.
2) P.L. Grande et al., Nucl. Instrum. Meth.,
B61 (1991), 282.
3) E. Friedland et al., Nucl. Instrum. Meth.,
B136-138 (1998), 147.
4) J.F. Ziegler, J.P. Biersack, and U.
Littmark, “The stopping and range of ions in solids,” New York: Pergamon, 1985.
5) M.W. Schmidt, K.K. Baldridge, J.A.
Boatz., J. Comp. Chem., 14 (1993), 1347.
6) T. Nakajima, K. Hirao, Chem. Phys. Lett.,
302 (1999), 383.
7) S. Huzinaga and B. Miguel, Chem. Phys.
Lett., 175 (1990), 289.
8) S. Huzinaga and M. Klobukowski, Chem.
Phys. Lett., 212 (1993), 260.
9) J. Kobus, L. Laaksonen, D. Sundholm,
Comput. Phys. Comm., 98 (1996), 346.
10) D.J. Land, J.G. Brennan, Atomic Data and
Nucl. Data Tables, 22 (1978), 235.