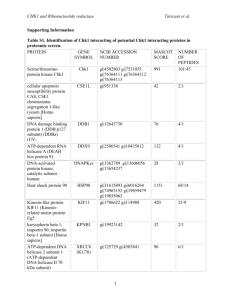
Supplementary Materials for: Metabolic interdependencies between
advertisement

Supplementary Materials for: Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer Kelly C. Wrighton, Cindy J. Castelle, Michael J. Wilkins, Laura A. Hug, Itai Sharon, Brian C. Thomas, Kim M. Handley, Sean Mullin, Carrie D. Nicora, Andrea Singh, Mary S. Lipton, Philip E. Long, Kenneth H. Williams, Jillian F. Banfield* * Corresponding Author: jbanfield@berkeley.edu This PDF file includes: I. Supplementary Text and Methods Genomic Binning Phylogenetic Analyses Genome Bins ACD17 and ACD79 Open-access data sharing Metabolic analyses Changes in relative abundance across the 10-day acetate stimulation CRISPR/CAS autoimmunity II. Supplementary Figures S1-S3, S6-S10 III. Supplementary Tables S4-S5 IV. Captions for Additional Supplementary Materials V. References for Supplementary Materials VI. Additional (separate documents) Supplementary Materials for this manuscript Figure S4 Figure S5 Table S1 Table S2 Table S3 FASTA file for Figures S8 and S9 1 I. Supplementary Text and Methods Genomic Binning. We first used tetranucleotide signal to cluster genomic fragments via ESOM (main text). For genome fragments > 10 kb, it was possible for 5 kb segments to fall into more than one cluster, especially if segment ESOM positions fell close to cluster boundaries. Fragments were only binned ‘confidently’ if > 50% of segments deriving from a genome fragment fell into the same cluster. In addition to tetranucleotide information, we used abundance changes over time to inform genomic binning. We computed the fraction of reads contributing to each contig from each dataset (A, C, and D) separately for all genome fragments > 5 kb (read abundances were normalized to account for differences in the sizes of the A, C and D genomic datasets). We projected the abundance ratio information ([A]/[D]) onto the ESOM to further differentiate ESOM clusters. The set of genome segments comprising each genome bin was exported by manually defining the boundaries in the ESOM and the region assigned an “ACD” cluster number. After completing ESOM binning as detailed in the main text methods, we were left with three sets of contigs unbinned: (i) those of 10 – 15 kb (represented by exactly 2 segments), where the two segments fell into different bins, and (ii) other fragments that had no bin assignment because they did not meet the >50% requirement for other reasons (e.g., when fragments fell into multiple different map regions) (iii) scaffolds < 5 kb. In case (i), fragments were assigned to one of the two bins randomly, but designated “p” (e.g., assigned as ACD11p). Many of these were checked to rule out obvious discrepancies in GC content and coverage. Scaffolds of type (ii) were named ACDUNK (ACD unknown) and all larger fragments (> 20 kb) were binned after genomic annotation via phylogenetic analysis and based on coverage (for very high coverage scaffolds). Some ACDUNK scaffolds were assigned to phylum-level groups (e.g., ACDOP11), others to ACDNov if no phylogenetic classification was possible. Scaffolds in case (iii) were binned by projection onto the ESOM map using their tetranucleotide frequency information. A histogram of sequence length and abundance ratio in the first (A) and last samples (D) shows well-resolved peaks, indicating differential abundance patterns of genomes across experimental time (Figure S2B). Peaks in the histogram were used to assign a color (representing the abundance ratio in samples A/D) to each data point in the ESOM (Figure S2C). In some cases (e.g., clusters 2 and 3), temporal abundance information assisted with the separation of adjacent clusters of fragments with similar tetranucleotide sequence composition. In total, 87 genome bins were identified (Figure S2D), while the remaining unbinned genome fragments were assigned to 13 bins of uncertain, but putative identification. This data is available on the ACD ggKbase website (see data sharing below). Phylogenetic analyses. For the concatenated 16 ribosomal proteins, each individual gene dataset was aligned using MUSCLE version 3.8.31 (Edgar RC, 2004) and then manually curated to remove end gaps and single-taxon insertions. Model selection for evolutionary analysis was determined using ProtTest3 (Darriba et al., 2011) for each single gene alignment. The curated alignments were concatenated to form a 16-gene, 729 taxa alignment with 3,544 unambiguously aligned positions. A maximum likelihood phylogeny for the concatenated alignment was conducted using PhyML (Guindon et al., 2003) under the LG+α+γ model of evolution and with 100 bootstrap replicates. FASTA of all nucleotide and protein sequences in the genomes: http://ggkbase.berkeley.edu/genome_summaries/88-ISME_Wrighton_GenomeCompletion For single protein trees (e.g NiFe and FeFe hydrogenase), the phylogenetic pipeline was performed as previously described (Wrighton et al., 2012). Briefly protein sequences were aligned using MUSCLE version 3.8.31 with default settings (Edgar RC, 2004). Problematic regions of the alignment were removed using very liberal curation standards according to (Sassera et al., 2011) with the program 2 GBlocks (Talavera et al., 2007). Alignments with and without GBlocks were confirmed manually. Best models of amino acid substitution for each protein alignment were estimated using ProtTest3 (Darriba et al., 2011). Phylogenetic trees were generated using RAxML (Stamatakis A, 2006) with the PROTCAT setting for the rate model and the best model of amino acid substitution specified by ProtTest. Nodal support was estimated based on 100 bootstrap replications using the rapid bootstrapping option implemented in RAxML. Genome Bins ACD17 and ACD79. One bin, ACD79, contained several genes of functional interest including genes for carbon utilization (Table S2) and NiFe hydrogenases (Figure S8), but contained only a few core genes for assigning phylogenetic affiliation (Figure S3). Moreover, phylogenetic genes that were present provided an inconsistent signal, indicating the bin was not representive of a single genome. For instance, many of the ribosomal proteins affiliated with Firmicutes (~16x coverage), while RecA (~20x coverage) was clearly associated with the phylum Chlamydiae. Coverage in the bin ranged from 3x to greater than 50x, consistent with a genomic bin composed of multiple genomes. Many of the key genes discussed in this manuscript are on small contigs less than five genes (e.g. cellobiose and NiFe hydrogenase), which do not have accurate phylogenetic markers for assigning these functions to a taxonomic identity. As such we conclude the ACD79 genomic bin contains genome fragments from several organisms; and assign these fragments to unknown phylogenetic affiliation. Another genomic bin (ACD17) also had a limited amount of marker genes, but those that were present (e.g. RpoB, S7, L3) had strong coherent phylogenetic affiliation with members of the Chlamydiae. Our proteomic data does suggest the presence and expression of Chlamydiae genes (from ACD17) in the aquifer. This finding was unexpected; given all members of the phylum Chlamydiae are obligate intracellular endosymbionts of Eukaryotes. When considering possible hosts, protozoan are likely candidates as the nearest related genome to ACD17 (Table 1) has protozoan hosts in other aquifer systems (Rolf et al., 2005), and recent 18S rRNA gene sequences collected from the Rifle aquifer revealed an abundant and diverse protozoan community (Holmes et al., 2013). Future research is necessary to untangle the obligate and facultative symbioses that may occur across the subsurface microbial community. Open-access data sharing. An overview of the genomes reconstructed (ACD1 – ACD87), showing the bin size, number of proteins predicted, average genome length/protein, phylogenetic affiliation (if known), average GC content, largest scaffold, and average coverage by read depth. The “abundance” estimate was determined based on genome coverage calculated from kmer coverage using the standard formula for interconverting these quantities. Note that these values may not estimate the true abundance of any organism in the community, because approximately 55.3% of the reads were not assembled during ACD genome assembly and some organisms have contigs <2 kb which may not be binned. Genomic bins can be accessed by clicking on the links for any of these ACD bins or by searching for specific genes in the search tool bar. After signing in users can create “lists” with text, EC number, or sequences (later via BLAST) as search terms. Additionally, these list features can be summarized using “genome summary” feature, which can provide access to DNA and amino acid sequence FASTA files. The binned data is included on the website: http://ggkbase.berkeley.edu/Rifle_ACD/organisms Metabolic analyses. While we previously identified the capacity for fermentation in at least five candidate phyla (SR1, WWE3, OD1, OP11, BD1-5, PER), our prior analyses did not include detailed summary of the carbon degradation capacity across these genomes (Wrighton et al., 2012). Here we identified glycoside hydrolases of selected functional classes (e.g. chitin, cellulase, debranching) by pfam HMM from all the 3 ACD bins. We then parsed this list for those that had high sequence similarity to a specific EC number. The remaining sequences, which could not be assigned to an EC number, were left with a putative designation based on pfam HMM classification. Both EC- and pfam-binned sequences are accounted for in Table S2, while Figure 3 summarizes the EC and pfam data into one summary value. The column entitled ‘Genome Phylogenetic Assignment’ includes taxonomic assignment and totals for each bin. All DNA and protein sequence data supporting GH analysis can be accessed via a genome summary on the website: http://ggkbase.berkeley.edu/genome_summaries/80-ISME_Wrighton_CarbonDegradationProfile We also examined genes for central carbon metabolism and the possible generation of fermentation end-products. First we examined genomes for presence of genes for glycolysis and TCA cycle. We then examined enzymes for converting pyruvate to acetyl-CoA via pyruvate dehydrogenase, pyruvate-formate lyase, and pyruvate ferredoxin oxidoreductase. Also included are genes for possible conversion of acetyl-CoA to ethanol, lactate, acetate, and butyrate (summarized in main text). We also highlight potential FeFe hydrogenases and NiFe hydrogenases, but note Figures S8 and S9 are based on manually curated data from these lists. We recognize many of these processes are reversible and only attempt to identify directionality in our near-complete genomic bins that lack evidence for respiratory metabolism (e.g. complete TCA cycle, pyruvate dehydrogenase, and electron transport machinery including complex 1). Of the carbon cycling organisms shown in Figure 5, we could only recover oxygen reductase genes from ACD77, a member of the Bacteroidetes. This genome has components of an aerobic respiratory chain including a cytochrome bd-type oxidase (ACD77_C00106G00003 and ACD77_C00106G00004). This bd-type terminal oxidase is composed by two subunits: subunit I and subunit II and contains three prosthetic hemes. The two genes identified in ACD77 are homologous to those of CydA (subunit I) and CydB (subunit II), respectively, of the cytochrome bd-type quinol oxidases from E. coli (42 and 26% identities, respectively). The heme-only cytochrome bd-oxidase is associated with microaerobic oxygen respiration (Richter et al., 2003). However, the this complex is also found in strictly anaerobic microorganism such Moorella thermoacetica, where it is been protects against oxidative stress and thus contributes to the limited O2 tolerance of M. thermoacetica (Amaresh et al., 2005). ACD77 has a partial NADH dehydrogenase (subunits D, E, F, C, B, A are present). However, given the partial nature of this genome, many required subunits (M, N, L involved in protons translocation and J, K, H, I and G) were not recovered. Our analyses recovered five FeFe hydrogenases in the ACD dataset, three from ACD20 (novel phylum) and two from ACD77 (Bacteroidetes) (Figure S7). Of the three confurcating hydrogenases in ACD20, ACD20_18461 has all the necessary residues for functionality (L1, L2, and L3 motifs). ACD20_9246_G0007, ACD20_9246_G0010, ACD77_9475.354011, and ACD77_9475.354011 contain all three motifs but have a replacement of a serine by cysteine in motif 1 (TSCSPGW rather than TSCCPAW), have a conserved replacement in motif 2 (MPCTAKFFE rather than MPCTAKKAE), and ACD20_9246_G0010 has a replaced cysteine in motif 3 with isoleucine. Phylogenetic analyses based on reference sequences from Schmidt et al. (2010) and recent database searches confirmed that the hydrogenase sequences were most closely related (58-72% AAI) to those from fermentative organisms (most obligately) including members of the Bacteroidetes (e.g. Anaerophaga thermohalophila), Clostridium spp., and Spirochaetes (Figure S7). Notably, two hydrogenase clusters, located on ACD20 contig 9246 and ACD77 contig 9475, share synteny and similarity, with the large subunit sequences from ACD77 and ACD20 more similar to each other (70%), than they are to homologs encoded on their respective contigs (~45%) (Figure S7). Previously we reported on and modeled type 4 and type 3b NiFe hydrogenases from OD1 and OP11 (Wrighton et al., 2012). Here we continue that analysis across the remaining 29 non-CP ACD genomes. Our phylogenetic analyses include only those sequences whose large catalytic subunit sequences contained at least one amino acid motif for functionality and are at least 60% complete (less than 75% sequences denoted as partial on tree). Sequences deposited as part of this analysis are in red, 4 those reported previously in green (OP11) and purple (OD1) (Figure S9). Complete amino acid FASTA files for ACD and reference sequences used to generate Figure S8 and S9 are available as a supplemental file. Multi-heme cytochromes were identified by manual and automatic curation via conserved CXXCH heme domains. Predicted phylogenetic affiliation, total heme content, and predicted cellular localization from PSORTb are included in Table S3. Sulfur and nitrogen functional genes were identified by annotation then confirmed manually. DNA or protein sequences can be accessed by searching GGKB website for gene loci provided in Tables S3-S5. Given the expression of genes indicative of active sulfate-reduction across our three time points, we evaluated the functional likelihood of the dissimilatory sulfite reductase complex (DsrAB) via modeling (Figure S7). Only the medium coverage DsrAB operon was complete enough to model. The DsrA was most closely related to a sequence from D. psychrophila LVS4 (95% AAI) and the DsrB from an unknown sulfate reducing bacterial sequence recovered from a contaminated aquifer in China (86% AAI). Protein modeling using using Swiss-model (Bordoli et al., 2009) with Desulfomicrobium norvegicum (DsrAB 65% AAI) confirmed that the substrate binding site, all cysteines coordinating the [4Fe-4S] clusters, and siroheme cofactors are conserved (Figure S7). The remaining predicted DsrAB partial protein sequences have high similarity to DsrAB in physiologically confirmed sulfate reducing bacteria, suggesting the protein products are functional (Table S4). In addition to the nitrate reductases recovered from genomes closely related to organisms with physiological capabilities (ACD10 and ACD23), the capacity for nitrate reduction (narGHIJ) was also confirmed in ACD75 contigs assigned to the Desulfobulbaceae. While the NarG was a partial sequence, we could verify the five conserved catalytic residues and the homology to nitrate reductase in other sulfate reducers, with a highest similarity to NarG from Desulfobacterium autotrophicum HRM2 (78% AAI). However, D. autotrophicum has not been documented to use nitrate as a terminal electron acceptor (Brysch et al., 1987), so the relevance to ACD75 physiology, and sulfate-reducing bacteria in general, is currently unknown. Currently, only one other member of the Desulfobulbaceae, Desulfobulbus propionicus, is known to grow by the reduction of nitrate (with propionate and sulfite as electron donors) via a periplasmic nitrate reductase (Widdel and Pfenning, 1982; Greene et al., 2003). Changes in relative abundance across the 10-day acetate stimulation. Microbial community structure was determined for the initial sampling (5 days after acetate, light blue bar), middle sampling (7 days after acetate, black triangle) and last sampling (10 days after acetate, dark blue) using the coverage of the scaffold containing the single copy ribosomal protein S3 (Figure S6). Two organisms, Dechloromonas spp. (ACD10) and a member of the Desulfobulbaceae (ACD75), increase in abundance dramatically at the second time point, when the Fe(II) concentration is highest. Specific members of BD1-5 (ACD3 and ACD4), OD1-i (ACD1, 5, 8, 11), and Deltaproteobacteria (ACD75 and ACD53) increase in abundance with increasing amendment time, yet other members of these same taxonomic groups show a decrease in abundance with time (Figure S6). These findings suggest that certain taxa respond to acetate availability, however strong interdependencies would be hard to identify with these data alone. For example, some taxa may respond to increased abundance of acetate-respiring bacteria, yet themselves may not be able to utilize acetate. CRISPR/Cas Adaptive phage immunity. Given the recovery of phage genomic data, and the detection of expressed phage genes in situ (Table 1), we examined CRISPR/Cas adaptive phage immunity across the data set. We only detected evidence for two or more Cas proteins in one member of each of the OP11, OD1, and BD1-5 Candidate phyla, and in a small subset of other Bacteria (ACD10, ACD23, ACD34, ACD39, ACD47, ACD60, ACD62, ACD75, ACD79). Interestingly, the ACD79 mixed genome bin contains 25 Cas proteins. Previous estimates, based primarily on isolate genome surveys, suggest that about 40 % of bacteria have CRISPR loci (Jansen et al., 2002; Stern et al., 2010). The current study would suggest that these systems 5 are relatively underrepresented in the Candidate Phyla. Surprisingly, no spacer sequences extracted from the CRISPR loci matched (even imperfectly) to any phage or other sequence in the dataset, raising the possibility of other (possibly as yet unrecognized) defense mechanisms in these organisms. However, it is also possible that the detected phage may be targeting bacteria retained on the 1.2 µm filter whereas phage targeting the genomes sampled here may have passed through the 0.2 µm filter. 6 II. Supplementary Figures Figure S1. A. Groundwater was collected during secondary acetate stimulation, meaning the aquifer was also amended with acetate the year prior. Dotted red box in part A. is highlighted in B., which details the well geochemistry during sample collection. Metagenomic samples (A, C, D) were collected 5, 7, and 10 days from the start of secondary acetate amendment during iron reduction. 7 8 Figure S2. A. ESOM constructed using tetranucleotide frequency information for fragments > 5 kb in length. Boundaries (dark bands) separate regions (collections of genome fragments) with similar signatures (each 5 kb fragment appears as a dot). B. Abundance ratios of fragments >5 kb in the A and C samples from the time series. Specific abundance ratio ranges were assigned colors for application to the ESOM. C. Gradation of color (shown in B.) was used to code the abundance ratio for each fragment represented on the map to better define cluster boundaries. D. Discrete map regions were numbered (1 – 87, corresponding to ACD1 – ACD87). The red box shows the periodic repeat of the map. Unresolvable regions were not binned from the ESOM (see SOM). 9 Figure S3. Genome completeness estimates for all recovered genomic bins. Bins that contain >75% of the markers or more than one genome are included in part A, while part B contains the less complete genomes. This figure and the FASTA files that accompany it can be accessed here: http://ggkbase.berkeley.edu/genome_summaries/88-ISME_Wrighton_GenomeCompletion 10 11 Figure S6. Rank abundance profiles of ribosomal protein S3 (rpS3) defined phylotypes identified in the initial sample A (light blue bar chart), the middle sample C (triangle symbol), and last sample D (dark blue bar chart). The coverage of the rpS3-containing scaffold is plotted on the y-axis and the x-axis contains organisms organized left to right by their abundance in sample A. Taxonomic affiliation is noted by color symbol above the initial sample and below the rank abundance curve. Figure shows that low abundant organisms (e.g. ACD3, ACD1) in time point 1 become dominant in time point 3. 12 Figure S7. Maximum likelihood tree of FeFe hydrogenase large catalytic subunit sequences. The five ACD sequences from Bacteroidetes (ACD77) and a novel phylum (ACD20) are highlighted in red bold. Reference sequences are in black with nearest neighbors included based on BLAST against the NCBI nr database (May 2013). Bootstrap values (>50) are shown, based on 100 resamplings. 13 0.1 gi169257881 EDS71847 Anaerofustis stercorihominis DSM 17244 gi149905277 ABR36110 Clostridium beijerinckii NCIMB 8052 gi160624920 ABO425432 Clostridium butyricum gi488597 AAA85785 Clostridium saccharobutylicum gi50344697dbjBAD29951 Clostridium paraputrificum gi169296709 EDS78838 Clostridium perfringens 80 56 gi188498603 ACD51739 Clostridium botulinum E3 str Alaska E43 54 gi118134934 ABK61978 Clostridium novyi NT gi144836 AAA23248 Clostridium pasteurianum 61 gi557064 AAB03723 Clostridium acetobutylicum ATCC 824 100 gi146347733 EDK34269 Clostridium kluyveri DSM 555 99 gi209170690 ACI42788 Clostridium tyrobutyricum gi150272145 EDM99349 Pseudoflavonifractor capillosus ATCC 29799 gi167665067 EDS09197 Anaerotruncus colihominis DSM 17241 gi167656200 EDS00330 Eubacterium siraeum DSM 15702 gi167660893 EDS05023 Clostridium scindens ATCC 35704 gi160430589 ABX44152 Clostridium phytofermentans ISDg gi158440937 EDP18661 Clostridium bolteae ATCC BAA 613 gi145848527 EDK25445 Ruminococcus torques ATCC 27756 gi210149924 EEA80933 Clostridium nexile DSM 1787 gi158140165 ABW18477 Alkaliphilus oremlandii OhILAs gi145410445 ABP67449 Caldicellulosiruptor saccharolyticus DSM 8903 gi125713088 ABN51580 Clostridium thermocellum ATCC 27405 gi219999973 ACL76574 Clostridium cellulolyticum H10 gi149951660 ABR50188 Alkaliphilus metalliredigens QYMF gi14250935embCAC39231 Eubacterium acidaminophilum gi167704730 EDS19309 Clostridium ramosum DSM 1402 100 gi169292311 EDS74444 Clostridium spiroforme DSM 1552 gi169247661 ACA51661 Thermoanaerobacterium saccharolyticum JW/SL YS485 gi206738217 ACI17295 Coprothermobacter proteolyticus DSM 5265 gi214035763 EEB76457 Carboxydibrachium pacificum DSM 12653 70 85 gi166855001 ABY93410 Thermoanaerobacter sp X514 gi166856698 ABY95106 Thermoanaerobacter pseudethanolicus ATCC 33223 gi158440918 EDP18642 Clostridium bolteae ATCC BAA 613 Spirochaeta smaragdinae DSM 11293 YP 003802215 85 88 100 97 73 88 57 78 97 90 68 100 74 ACD20 9246704834G0007 Phaeospirillum molischianum ACD77 9475354011G0004 75 100 95 100 100 57 65 51 88 65 98 52 78 100 95 90 100 DSM 120 ZP 09877549 Anaerophaga thermohalophila DSM 12881 ZP 08845406 gi134052208 ABO50179 Desulfotomaculum reducens MI 1 gi121307228 EAX48145 Thermosinus carboxydivorans Nor1 gi83573467 ABC20019 Moorella thermoacetica ATCC 39073 gi169638340 ACA59846 Desulforudis audaxviator MP104C gi206743093 ACI22150 Thermodesulfovibrio yellowstonii DSM 11347 gi169637214 ACA58720 Desulforudis audaxviator MP104C gi85722031 ABC76974 Syntrophus aciditrophicus SB gi114338877 ABI69725 Syntrophomonas wolfei Pelotomaculum thermopropionicum strain SI YP 001212560 gi146274442dbjBAF60191 Pelotomaculum thermopropionicum SI gi134053819 ABO51790 Desulfotomaculum reducens MI 1 gi149948338 ABR46866 Alkaliphilus metalliredigens QYMF gi118133456 ABK60500 Clostridium novyi NT gi187773177 EDU36979 Clostridium sporogenes ATCC 15579 gi160359833 ABX31447 Petrotoga mobilis SJ95 gi212676039 EEB35646 Anaerococcus hydrogenalis DSM 7454 gi115252467embCAJ70310 Clostridium difficile 630 100 gi164602497 EDQ95962 Clostridium bartlettii DSM 16795 100 gi164604047 EDQ97512 Clostridium bartlettii DSM 16795 gi160428861 ABX42424 Clostridium phytofermentans IS 100 gi164604201 EDQ97666 Clostridium bartlettii DSM 1 85 gi188500220 ACD53356 Clostridium botulinum E3 str 96 gi149903112 ABR33945 Clostridium beijerinckii NCIM gi167660485 EDS04615 hydrogenase Fe only Alistipes putredinis DSM 17216 gi149936113 ABR42810 Parabacteroides distasonis ATCC 8503 100 99 gi218224548 EEC97198 Parabacteroides johnsonii DSM 18315 gi154085279 EDN84324 Parabacteroides merdae ATCC 43184 gi77545287 ABA88849 poss Pelobacter carbinolicus DSM 2380 100 gi77545315 ABA88877 poss Pelobacter carbinolicus DSM 2380 79 gi466366 AAA87057 Desulfovibrio fructosovorans gi189435405 EDV04390 Bacteroides intestinalis DSM 17393 100 gi29337426 AAO75231 Bacteroides thetaiotaomicron VPI 5482 73 95 gi149129481 EDM20695 Bacteroides caccae ATCC 43185 gi156107991 EDO09736 Bacteroides ovatus ATCC 8483 Symbiobacterium thermophilum IAM14863 BAD42191 gi51858033dbjBAD42191 Symbiobacterium thermophilum IAM 14863 gi169638544 ACA60050 Desulforudis audaxviator MP104C gi146273809dbjBAF59558 Pelotomaculum thermopropionicum SI gi167591797 ABZ83545 Heliobacterium modesticaldum Ice1 100 100 gi94448908embCAJ44289 Heliobacillus mobilis Heliobacillus mobilis CAJ44289 gi219992456 ACL69059 Halothermothrix orenii H 168 gi160360253 ABX31867 Petrotoga mobilis SJ95 ACD77 9475354011G0001 ACD20 9246 70483 14G0010 gi177840576 ACB74828 Opitutus terrae PB90 1 gi186971103 ACC98088 Elusimicrobium minutum Pei191 gi57225451 AAW40508 poss Dehalococcoides ethenogenes 195 100gi73659815embCAI82422 poss Dehalococcoides sp CBDB1 gi146269821 ABQ16813 Dehalococcoides sp BAV1 gi89336906dbjBAE86501 poss Desulfitobacterium hafniense Y51 98 gi134051038 ABO49009 Desulfotomaculum reducens MI 1 65 gi146273100dbjBAF58849 Pelotomaculum thermopropionicum SI 87 gi169637216 ACA58722 Candidatus Desulforudis audaxviator MP104C gi6650985 AAF22114 Megasphaera elsdenii DSM 20460 100 gi158434701 EDP12468 Clostridium bolteae ATCC BAA 613 88 gi167662588 EDS06718 Clostridium scindens ATCC 35704 81 gi150274152 EDN01243 Pseudoflavonifractor capillosus ATCC 29799 85 gi167654850 EDR98979 Anaerostipes caccae DSM 14662 gi169297952 EDS80043 Clostridium perfringens gi149905080 ABR35913 Clostridium beijerinckii NCIMB 8052 70 gi146346356 EDK32892 Clostridium kluyveri DSM 555 gi115252370embCAJ70211 Clostridium difficile 630 85 100 gi164604064 EDQ97529 Clostridium bartlettii DSM 16795 gi51858116dbjBAD42274 Symbiobacterium thermophilum IAM 14863 gi83573631 ABC20183 Moorella thermoacetica ATCC 39073 gi50875368embCAG35208 poss Desulfotalea psychrophila LSv54 93 gi78217927 ABB37276 Desulfovibrio alaskensis G20 gi114338374 ABI69222 Syntrophomonas wolfei subsp wolfei str Goettingen gi116697357 ABK16545 Syntrophobacter fumaroxidans MPOB 100 67 gi1914864embCAA72423 Desulfovibrio fructosovorans JJ 100 gi13022069 AAK11625AF331719 Desulfovibrio alasken 77 78 gi46449596 AAS96246 Desulfovibrio vulgaris Hildenboroug gi206741320 ACI20377 Thermodesulfovibrio yellowstonii DS 100 gi167353361 ABZ75974 Shewanella halifaxensis HAW EB4 100 gi24350246 AAN56895 Shewanella oneidensis MR 1 85 70gi117611495 ABK46949 Shewanella sp ANA 3 gi88660702 ABD48098 Shewanella decolorationis 85 gi113886266 ABI40318 Shewanella sp MR 4 gi187438956 ACD10930 Blastocystis sp NandII gi149793997 ABR31445 Thermosipho melanesiensis BI429 100 gi154152871 ABS60103 Fervidobacterium nodosum Rt17 B1 92 gi157314808 ABV33907 Thermotoga lettingae TMO 53 gi147735403 ABQ46743 Thermotoga petrophila RKU 1 100 gi170176047 ACB09099 Thermotoga sp RQ2 gi115519803 ABJ07787 Rhodopseudomonas palustris BisA53 100 gi46449598 AAS96248 Desulfovibrio vulgaris Hildenborough gi217991372 EEC57378 Bacteroides pectinophilus ATCC 43243 gi188498811 ACD51947 Clostridium botulinum E3 str Alaska E43 87 100 gi210154580 EEA85586 Clostridium hiranonis DSM 13275 55 gi167655182 EDR99311 Anaerostipes caccae DSM 14662 91 gi167710039 EDS20618 Clostridium sp SS2/1 100 gi41817525 AAS12110 Treponema denticola ATCC 35405 93 gi166028598 EDR47355 Dorea formicigenerans ATCC 27755 58 gi167662445 EDS06575 Clostridium scindens ATCC 35704 67 gi210152607 EEA83613 Clostridium nexile DSM 1787 gi149752498 EDM62429 Dorea longicatena DSM 13814 gi134052211 ABO50182 Desulfotomaculum reducens MI 1 gi149792909 ABR30357 Thermosipho melanesiensis BI429 66 gi154152860 ABS60092 Fervidobacterium nodosum Rt17 B1 100 gi217336057 ACK41850 Dictyoglomus turgidum DSM 6724 72 gi157314419 ABV33518 Thermotoga lettingae TMO 68 gi221572431 ACM23243 Thermotoga neapolitana DSM 4359 100 96 gi147736041 ABQ47381 Thermotoga petrophila RKU 1 gi4981990 AAD36496AE001794 Thermotoga maritima MSB8 gi170287484dbjBAG14005 Termite group 1 bacterium phylotype Rs D17 100 gi118502624 ABK99106 Pelobacter propionicus DSM 2379 100 100 64 74 90 100 59 68 100 ACD20 18461244044G0012 73 74 100 100 87 100 100 99 100 100 73 80 gi156864730 EDO58161 Clostridium sp L2 50 gi167653849 EDR97978 Anaerostipes caccae DSM 14662 gi153794181 EDN76601 Ruminococcus gnavus ATCC 29149 gi149831165 EDM86254 Ruminococcus obeum ATCC 29174 gi166027777 EDR46534 Dorea formicigenerans ATCC 27755 gi158434681 EDP12448 Clostridium bolteae ATCC BAA 613 gi158140225 ABW18537 Alkaliphilus oremlandii OhILAs gi125713176 ABN51668 Clostridium thermocellum ATCC 27405 gi56387327 AAV86076 Clostridium saccharoperbutylacetonicum ATCC 27021 gi160426914 ABX40477 Clostridium phytofermentans ISDg gi182378378 EDT75909 Clostridium butyricum 5521 gi149905387 ABR36220 Clostridium beijerinckii NCIMB 8052 gi188498443 ACD51579 Clostridium botulinum E3 str Alaska E43 14 72 100 Grp3 52 76 100 85 56 mo na lo ro ch De Grp3c putative YP001717784 Desulforudis audaxvia 79 Grp3c Deha lococcoides Grp3c ethenogenes a NP YP 848 195 6135 058 Syn Grp3 53 c YP Grp3 tropho 0647 bacter a Me 4Des Q0thanop fumaro ulfo xidans Gr Grp3 0404 yrus tale MPO p3 c kand Met a ps c Y ychr NP P447 hanoc leri AV ophi 27 occ la LS 19 62 369 us v54 Met 62 vol han tae Me th os ph a an ot era YP h e 0 rm sta YP03 ob dtm ac 0052 an te Gr ae 3842 r p3 DSM th 4731 er 3 6 Si AC d A 30 ma 91 6 de D1 AD3 ut ot 80 Ga ro 0 ro 65 8 l l xy ph R ic i o d a 91 us 97 hodo ne ns De ba ll l i lt H ct a th 25 a e r 51 ca ot ca ps ro ps 6G ul i f ph 00 at er icu u 01 s ri s fo ES p 1 rm ar an ti s al ES 2 eobacteria ACD62 30930 12218 13G0001 Deltaprot xi 546 7078 ACD34 223 fle 05 tial Chloro terium MLMS1 9 N bac si 7G0006 par 53 teo abys 1 pro AB hrix ed OP3 7 Delta GC ldit 129175 SC ltur 00 81 Ca ve ZP0 Uncu 6G0 eon 5509 ZP09 5558 81 1 rcha BAL5 3 6 le s a tive a 0 88 at 113 oplasm rm The A CD tive ta c pu 100 70 3C 92 B B1 RC AM a ic um at ic 12 om et H6 gn ar ma um m il xi o ph lu le om mo il er of or ir th or hl osp l c s mu Ch De net 1 lo g 8 3 U NI og Ma 20 00 la ty 84 759 G0 mophi ic 4 2 9 D r 4 LSv5 YP 22 the 97 59 la d YP4 50 inea 51 phi p3 d 002 22 chro PCC7 Grrp3 95 erol 00 psy sp G YP 876 Ana ea cus 00 ve ococ otal ti 34 736 ulf nech 02 ta CD 41 Des 469 Sy 24 75 s DSM110 Pu A YP00 948 PCC ovoran 1733 d 65 peptid p3 rp3d YP 0 d YP002 Nostoc io r ibr G G d Grp3 7520 sulfov Grp3 d YP0070 7 Dethio DSM13181 639310 m mobile Grp3 ve ZP0 Anaerobaculu 594 Putati 6444 3d Grp d Putative YP00 BAA1850 Grp3 Anaerobaculum hydrogeniformans Grp3d Putative ZP06439013 s na 100 85 98 68 95 78 53 3D 59 9777 100 98 79 62 ACD22 38087 6568 19G0008 OP11 004 OP11 28 5419 7G0 ACD57 914 92 3B 53 92 70 100 99 63 100 60 95 AH T 1 47 75 i DSM113 ss stonii by 180 ix a o yellow ovibri DSM ithr us SB a desulf phic sum Thermo ld ri itro 49644 vino 2 Caea acid te YP0022 phus Grp3b ialomatium 69269i8amat bac D1i ntro t Sy r o O t pa lochr 0173 00 WP dus YP46 te 004 ro 01 1G0 2786 Al ab rh 4 2 a p 00 44 lo 319 003 mm 7G Ha YP 11 4 5 Ga 74 371 e3b 66 a 23 tiv 52 22 08 ri DG1 0 08 0 Puta 0 0 4 WP G te m 8 10 ac r i u 28 74 ob cte 9 1 e D 9 t ba 6 AC ro e 55 ap bia 35 mm r o 1 D2 Ga omic AC 01 rruc 0 0 e 6G 60 V 62 43 05 43 05 4 ZP 59 b 6 3 51 ve ti 46 ta Pu CD 4 56 ct e r al ka li p hi l us 1 th i ob a 73 83 99 0 De 100 72 8 51 70 56 99 52 71 100 Gr 100 9 57 50 91 CC AT ca ri te en ns la ma el or um of on br en lm og ru Sa dr m 1 lu s hy il 64 u 38 ir erm 85 36 h sp tei V7 t a o M ng od xydo AA DS hu Rh o b um p4 us 11 Car ll Gr os ri 12 i lu m p ri 45 64 7 os cel fu AC 60 an A 3 rmo th 1 us 4 YP the Me BAV rp a cc 6 um G 8 sp p4 co idi 31 Gr des 50 ro str coi YP lo Py ococ 1 C p4 hal gas Gr 409 39 o gi 8 De 010 736 ibri lfov YP0 ABQ1 4 Desu Grp Grp4 1029 AAP5 Grp4 3 16 65 74 100 83 p4 Pu 100 ACD14 Gr EEB ta PutativeGrp4 27 3422 42G0005 OD1i Pu 100 ti p4 73 ta YP 113608 4 ve ACD5 75 ti Puta Methylococcu E E 25 Gr ve 65 s capsulat tiv 24 3130 Gr p4 B7 T eGr p4 9G0003 29 her ZP p4 CA mo Pu ACD ACD7 12 91 YP OD 09 J7 t c 0 9 a 25 3 14 1i 65 oc tiv 0379 Gr T 362 23 29 cu 940 eG 984 p4 her 0 9 2 r C s 9 77 p4 6 an 171 Can G001 sp CA moc De di NC1 57 did 1 OD da su oc A3 0 9 at A G00 1i tu lf M4 bac us cu 55 s 04 os Nitr ter s Ku 50 po OD1 iu osp en sp ro 1 i m D en ira si ia utc Es AM def nu h st ch 4 luv s sed ut ii er yo tg ime un ic ar nt gi ti hi ae e ns a is DS co M li 17 73 4 100 NP 5 58 77 p4 71 65 Gr AC D7 100 51 100 3 2941 ATCC ilis riab na va abae 73102 me PCC 87 An ctifor 3250 a YP toc pun Grp2 77 Nos AAC162 Grp2a 5 100 8882 opersicina capsa rose 0740 Thio um japonicum USDA110 Grp2b GAAX4 rp2b NP773583 Bradyrhizobi 35 21 19 Gr 99 p1 6 YP 28 7 AC 06 91 2 9 D5 43 11 3 32 G 0 0 27 De G0 01 su 79 AC D l 3 00 f Gr ot 74 p 30 p1 6 97 YP0 16 alea 1 D art 01 67 e p i 24 229 24 6 G0 sych sul al 652 9 1 0 ro fo D Ge ph 0 ob a 0 1 G b e Grp1 00 0 i ZP07 2 p cter u Geob la L ulbsul 3354 art ran ac Sv5 ac fo 71 ial iire te 4 ae b Desu lfov a ul pladucer ibri ba sm ns o fr ca ucto id Rf4 so ea Grp1 vora 92 89 AC D7 A YP00 ns 3329 JJ 621 Grp1 YP3 Deha loco 60377 ccoi Carbox des ydothe spVS rmus hyd rogeno forman sZ2901 Grp2b YP00 2526186 Rhod obacter spha eroides KD13 1 Grp2b AAC32033 Rhodobacter capsulatus at i ve 3 b ZP 03 98 Pu t 79 puta 70 72 9 EMR ve 100 93 100 89 ACD Grp3 GE5 D1 4070736 Ther ios mococcusus DSM363 barophil8us 53 MP NP 579061 hydrogenaseII alpha Pyrococcus furiosus DSM3638 ati Grp3c put at i pu t A AC CD1 D9 1 12 1 2 D6 6 4 7 38 76 98 21 1 3 86 26 57 5 1 4 80 AC 479 9G5 D7 7 0 29 30 6G 013G0 0 2 4 69 004 OD77 04 O 1 i O AC D6 83 d1 D1 3 G0 i i 66 A CD 24 00 99 55 83 7 74 4 351 76 OD 43 80 8 97 1i 71 5 7 52 96 9G A CD 5 6 0 58 G00 014 251 Grp3 21 O 05 b YP 1844 63 6 OD 1 D 1 82 Th 38 ermo Grp3b 1 1G cocc NP1265 Grp3b us 001 NP5786 48 Pyr ko 23 Pyr ococcu dakare 2 O ococcu s abyssi nsis Grp3b YP00 D1 s fur KO AC c Grp3 3c Grp 1i i ODODi1 1 08 00 04OD 8G G006 i 1 00 i D 1 47 91 65 9 G0 OD1 O 4 2 74 1 6 15 99 49 0 8 78 00 00 9 0 G 4 6 D1 K6 15 9G 9 AC UN 3 4 3 5 1 D1 756 677 43 AC 8 17 6 29 1 7 D5 AC D5 99 AC 8 D AC 0.1 s Figure S8. Maximum likelihood tree of NiFe hydrogenase sequences with hydrogenase class groupings (3B, 3C, 3D, 1, and 4) that contain ACD sequences highlighted in bold. ACD sequences previously reported are colored purple (OD1) and green (OP11). ACD sequences reported as part of this manuscript are in red. Reference sequences are in black and include those that have been physiologically confirmed and nearest neighbors that lack physiological confirmation (noted as putative). Bootstrap values (>50) are shown, based on 100 resamplings. 15 Figure S9. A) In silico modeling of DsrAB from ACD75 (~40x coverage fragment) to the known structure of Desulfomicrobium norvegicum (shown in B., PDB:2XSJ). The ACD75 medium-coverage DsrA and DsrB sequences have 66% and 65% amino acid identity with the corresponding subunits from D. norvegicum. The four [4Fe-4S] clusters from the complex DsrAB are colored in black and the two sirohemes are colored in red. 16 Figure S10. Proteomic expression of sulfate reduction genes across the three time points, with key genes expressed as early as five days after acetate stimulation (sample A). Three distinct copies of the putative sulfate reduction pathway were identified from scaffolds with high (>65X), medium (~40X), and low coverage (<10X). Grey box indicates the gene was not identified in the metagenome. All scaffolds were from the ACD75 bin and located on contigs with closest relationship to members of the Desulfobulbaceae. 17 III. Supplementary Tables Table S4. Dissimilatory sulfite reductase genes identified in the ACD metagenome. Gene ID Subunits Length (aa) % of identity % of identity % of identity Best hits in NCBI (blastP) Desulfomicrobium Archaeoglobus fulgidus (PDB: 3MMC) Desulfovibrio vulgaris (PDB: 2V4J) % of identity Norvegicum (PDB: 2XSJ) ACD75_2650.24017.53 G0028 Alpha subunit 117 38 45 38 71% Chlorobium (YP_910622) ACD75_7481.6912.26G 0006 Alpha subunit 80 38 40 33 64% Pelodictyon (YP_002019133) ACD75_7481.6912.26G 0005 Alpha subunit 118 41 47 43 66% Chlorobium (YP_001960255) ACD75_7481.6912.26G 0007 Beta subunit 144 36 44 40 76% Prosthecochloris (YP_002014743) ACD75_2265.5038.64G 0001 Alpha subunit 151 61 49 62 93% uncultured prokaryote (AEZ49823) ACD75_2265.5038.64G 0002 Beta subunit 375 64 54 63 87% Desulfotalea (YP_064534) ACD75_5279.2885.33G 0001 Alpha subunit 216 39 45 39 64% Chlorobium (YP_001960255) phaeobacteroides ACD75_5279.2885.33G 0002 Beta subunit 358 42 47 45 75% Chlorobium (YP_910623) phaeobacteroides ACD75_142.16489.41G 0013 Beta subunit 375 65 55 63 86% Desulfotalea (YP_064534) ACD75_142.16489.41G 0014 Alpha subunit 349 66 53 66 95% Uncultured sulfate bacterium (ABK90687) 18 phaeobacteroides phaeoclathratiforme phaeobacteroides aestuarii psychrophila psychrophila reducing Table S5. Key functional genes for relevant nitrogen metabolisms identified in the ACD metagenome. Genes greater than 75% of alignment length are denoted as complete, less than 75% are partial. Organism Dechloromonas ACD10 Dechloromonas ACD10 Dechloromonas ACD10 Dechloromonas ACD10 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Comamonadaceae ACD23 Desulfobulbacaea ACD75 Desulfobulbacaea ACD75 Desulfobulbacaea ACD75 Best hit against NR NCBI database (including percentage of identity, annotation, Amino amino acid length and accession number) acid length ACD10 Dechloromonas: Nitrate and Nitrite Reduction ACD10_59995.11181.12G0004 cytochrome c-type 161 62% protein NapB complete Candidatus Accumulibacter phosphatis clade IIA str. UW-1 Nitrate reductase cytochrome c-type subunit (NapB) (155aa) (YP_003169088) ACD10_C00026G00001 periplasmic nitrate 365 89% reductase NapA partial Dechloromonas aromatica RCB (periplasmic nitrate reductase subunit NapA) (837aa) (YP_286714) ACD10_19428.2894.19G0002 cytochrome d1, heme 561 93% region complete Dechloromonas aromatica RCB (cytochrome d1, heme region)(561aa) (YP_286474) ACD10_3191.2771.13G0001 cytochrome c, class 611 88% I:cytochrome d1, complete Dechloromonas aromatica RCB (cytochrome c, class I:cytochrome d1, heme region) heme region (576aa) (YP_286522) ACD23, a member of the Comamonadaceae: Nitrate, Nitrite, and Nitrous-oxide Reduction ACD23_30478.2373.10G0001 nitrate reductase, 35 83% alpha subunit partial Rhodoferax ferrireducens T118 (respiratory nitrate reductase alpha subunit) (1272aa) (YP_524035) ACD23_112229.2031.9G0004 Nitrate reductase, 38 97% alpha subunit partial Acidovorax sp. KKS102 (nitrate reductase subunit alpha) (1265aa) (YP_006852832) ACD23_120092.2114.10G0002 nitrate reductase 1, 166 100% beta subunit partial Acidovorax radices (nitrate reductase A subunit beta) (507aa) (WP_010463003) ACD23_120092.2114.10G0001 nitrate reductase, beta 153 82% subunit partial Salmonella enterica (nitrate reductase subunit 2 beta) (547aa) (WP_006635366) ACD23_120092.2114.10G0003 nitrate reductase, beta 84 84% subunit partial Proteus penneri (hypothetical protein) (74aa) (WP_006535459) ACD23_120092.2114.10G0004 nitrate reductase 1, 244 98% alpha subunit partial Acidovorax delafieldii (nitrate reductase A subunit alpha) (1266aa) (WP_005795701) ACD23_65655.2560.11G0002 nitrite reductase (NO572 85% forming) complete Acidovorax sp. JS42 (nitrite reductase) (574aa) (YP_986168) ACD23_25640.2198.11G0002 nosZ; nitrous-oxide 568 92% reductase complete Acidovorax delafieldii (Nitrous-oxide reductase) (645aa) (WP_005797511) ACD23_65655.2560.11G0002 nitrite reductase (NO572 85% forming) complete Acidovorax sp. JS42 (nitrite reductase) (574aa) (YP_986168) ACD75, member of the Desulfobulbaceae: Potential for Nitrate and nitric-oxide reduction, and NrfA ACD75_7481.6912.26G0009 NarG, nitrate 219 69% reductase 1, alpha partial Desulfotignum phosphitoxidans (nitrate reductase alpha chain) (668aa) (WP_006968765) subunit ACD75_1208.4520.27G0007 NarG, nitrate 164 78% reductase 1, alpha partial Desulfotignum phosphitoxidans (nitrate reductase alpha chain) (561aa) (WP_006968766) subunit ACD75_2523.3723.38G0001 nitric-oxide reductase 393 44% partial Geobacillus kaustophilus HTA426 (nitric oxide reductase cytochrome b subunit) 9 Gene ID Primary annotation 19 Desulfobulbaceae ACD75 ACD75_11188.12436.33G0010 Desulfobulbaceae ACD75 ACD75_11188.12436.33G0011 Desulfobulbaceae? ACD75-small contig (could be misbin) yet high coverage is consistent with ACD75 ACD75_C00095G00010 Desulfobulbaceae ACD75 ACD75_2650.24017.53G0012 Geobacter ACD53 ACD53_41450.4833.18G0001 Geobacter ACD55 ACD55_37793.7867.12G0004 Geobacter ACD75-misbin moved to ACD55 ACD55_C00094G00007 Novel Deltaproteobacteria ACD73 Bacteroidales ACD77 ACD73_290737.2868.6G0004 ACD77_93884.135712.12G0038 hypothetical protein; nitric-oxide reductase, cytochrome bcontaining subunit I nitric-oxide reductase, cytochrome bcontaining subunit I K04561 nitric-oxide reductase, cytochrome bcontaining subunit I 395 partial nrfA; cytochrome c552 498 complete nrfA; cytochrome c552 496 complete (790aa) (YP_146611) 59% Desulfomonile tiedjei DSM 6799 (nitric oxide reductase large subunit) (758aa) (YP_006447466) 314 partial 70% Desulfomonile tiedjei DSM 6799 (nitric oxide reductase large subunit) (758aa) (YP_006447466) 111 partial 59% Geobacter daltonii FRC-32 (nitric-oxide reductase) (774aa) (YP_002535496) 83% Desulfobulbus propionicus DSM 2032 (respiratory nitrite reductase (cytochrome; ammonia-forming)) (498aa) (YP_004195416) Geobacter related Nitric-oxide reductase and NrfA cytochrome c nitrite 61 100% reductase, catalytic partial Geobacter sulfurreducens PCA (cytochrome c nitrite and sulfite reductase, catalytic subunit NrfA subunit lipoprotein) (490aa) (NP_954195) norZ; nitric oxide 763 100% reductase complete Geobacter bemidjiensis Bem (nitric oxide reductase (cytochrome b)) (763aa) (cytochrome b) (YP_002140689) cytochrome c; nitric110 77% oxide reductase, partial Geobacter bemidjiensis Bem (cytochrome c) (238aa) (YP_002138064) cytochrome ccontaining subunit II Other organisms nitrogen metabolism periplasmic nitrate 148 54% reductase NapA partial Maribacter sp. HTCC2170 (nitrate reductase catalytic subunit) (775aa) (YP_003861668) 20 69% Parabacteroides goldsteinii (cytochrome C nitrite reductase subunit c552) (494aa) (WP_007659437) IV. Captions for separate supplementary materials Figure S4. Complete maximum likelihood tree from Figure 2. The tree is based on sixteen ribosomal protein gene alignments concatenated and manually curated to form a 3,544-position, 729-taxon alignment. The ribosomal proteins used were rpL 2, 3, 4, 5, 6, 14, 15, 16, 18, 22, 24, and rpS3, 8, 10, 17, and 19. The best substitution model was determined for each alignment using ProtTest, and all alignments shared a common model of best fit. The complete concatenated tree was conducted using PhyML under the LG+I+γ model of evolution with 100 bootstrap resamplings. Sequences including the moniker “ACD” are from this dataset. Bootstrap support values of 50 or greater are included on the tree. Figure S5. Lineage specific concatenated ribosomal protein trees with reference sequences from JGIIMG (05/01/13) in the lineage. A) Alphaproteobacteria B) Betaproteobacteria C) Gammaproteobacteria D) Deltaproteobacteria E) Spirochaeta F) Bacteroidetes. Table S1. Summary of proteomic data for the A, C, and D samples (spectral count and normalized spectral abundance factor (NSAF) values are reported. Results for organisms from the same lineage are grouped, and within groups they are arranged approximately by protein abundance. Bold highlight flags proteins identified by two or more peptides. Table S2. Glycoside hydrolases (GH) identified by pfam domain/EC number in each of the ACD genomic bins. In the cases where organisms contained more than one GH type, it was noted by ACD bin number x and the number of GH identified (e.g. 77x2 denotes bin ACD77 has two GH). Parentheses within the table text sum the total number of GH affiliated with each phyla: OD1, OP11, BD1-5, ACD20 and ACD47 novel Phyla (NOV), WWE3, Proteobacteria (Prot), Firmicutes (Firm), Chloroflexi (Chloro), and GH in bins with unknown phylogenetic affiliation (UNKN). For some categories, no GH were not detected (ND). Table S3. Inventory of c-type cytochromes identified in the ACD metagenome including organism affiliation, predicted subcellular localization, and heme content. 21 V. References for Supplementary Materials Abascal F, Zardoya R, Posada D. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics, 21(9), 2104–2105. Brysch K, Schneider C, Fuchs G, Widdel F. (1987). Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Archives of Microbiology, 148(4), 264–274. Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. (2009). Protein structure homology modeling using SWISS-MODEL workspace. Nat protoc 4, 1-13. Darriba D, Taboada GL, Doallo R, Posada D. (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164-1165 Das A, Silaghi-Dumitrescu R, Ljungdahl LG, Kurtz DM. (2005). Cytochrome bd Oxidase, Oxidative Stress, and Dioxygen Tolerance of the Strictly Anaerobic Bacterium Moorella thermoacetica. Journal of bacteriology, 187(6), 2020–2029. Edgar RC. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. Greene EA, Hubert, C, Nemati M, Jenneman GE, Voordouw G. (2003). Nitrite reductase activity of sulphate‐ reducing bacteria prevents their inhibition by nitrate‐ reducing, sulphide‐ oxidizing bacteria. Environmental Microbiology, 5(7), 607–617. Holmes DE, Giloteaux L, Williams KH, Wrighton KC, Wilkins MJ, Thompson CA, et al. (2013). Enrichment of specific protozoan populations during in situ bioremediation of uranium-contaminated groundwater. The ISME journal, 1–13. Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1), 119. Jansen R, Embden JD, Gaastra W, Schouls LM (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Molecular microbiology, 43(6), 1565–1575. Michel R, MüLLER KD, Zoeller L, Walochnik J, Hartmann M, Schmid EN. (2005). Free-living amoebae serve as a host for the Chlamydia-like bacterium Simkania negevensis. Acta protozoologica, 44(2), 113–121. Richter OMH, Ludwig B. (2003). Cytochrome c oxidase: structure, function, and physiology of a redoxdriven molecular machine. Rev. Physiol. Biochem. Pharamacol. 147:47–74. Sassera, D, Lo N, Epis S, D'Auria G, Montagna M, Comandatore F., et al. (2011). Phylogenomic Evidence for the Presence of a Flagellum and cbb3 Oxidase in the Free-Living Mitochondrial Ancestor. Molecular Biology and Evolution, 28(12), 3285–3296. 22 Schmidt O, Drake HL, Horn MA (2010). Hitherto unknown [Fe-Fe]-hydrogenase gene diversity in anaerobes and anoxic enrichments from a moderately acidic fen. Applied and Environmental Microbiology, 76(6), 2027–2031. Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22(21), 2688–2690. Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends in Genetics, 26(8), 335–340. Talavera G, Castresana, J. (2007). Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Systematic Biology, 56(4), 564– 577. Widdel F, Pfennig N. (1982). Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids II. Incomplete oxidation of propionate by Desulfobulbus propionicus gen. nov., sp. nov. Archives of Microbiology, 131(4), 360–365. Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, et al. (2012). Fermentation, Hydrogen, and Sulfur Metabolism in Multiple Uncultivated Bacterial Phyla. Science, 337(6102), 1661–1665. 23