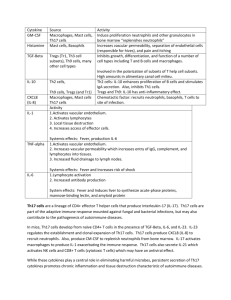
The capacity of retinoic-acid to induce phenotype
advertisement

Title
The capacity of retinoic-acid to induce phenotype-switching in differentiated Th17 Cells as a
control mechanism in immunological homeostasis
Summary
Clarifying the developmental relationship between immunosuppressive regulatory T cells
(Tregs) and inflammatory helper T 17 (Th17) cells is essential for the development of our
understanding of the delicate balance between homeostasis towards autoimmunity. However,
the interactions between these lineages remain incompletely elucidated. Recent work has
demonstrated considerable plasticity in differentiated Tregs, showing the capacity of T cells to
undergo phenotype-switching that involves the down-regulation of the expression of prototypical
Treg markers and the induction of the inflammatory Th17 phenotype. I seek to examine this
phenotype-switching in reverse: to determine the capacity of Vitamin A metabolite retinoic acid
to induce the Treg phenotype in differentiated Th17 cells and then to characterize the stability
and function of the resulting population. Although retinoic acid has been identified as a factor
that inhibits commitment to the Th17 lineage while supporting the expansion of the Treg subset,
its effects have been characterized only on unfractionated populations or naïve CD4+ T cells.
However, examining its effects on an isolated population of differentiated Th17 cells is of
considerable interest and relevance for the further elucidation of the relationship between the
Treg and Th17 lineages, and thus the balance between immunosuppression and autoimmunity.
Background and Significance
Effective immunological homeostasis relies on a continual balance between a number of factors,
including helper T (Th) cell activation and regulatory T cell (Treg) suppression. When
homeostasis is disrupted and the immune system tips in favor of activation, the host becomes
susceptible to autoimmunity. Although it was originally believed that many autoimmune
disorders, including rheumatoid arthritis (RA) and multiple sclerosis, partially resulted from an
aberrant Th1 response, several studies in mice that demonstrated the deleterious effects of
interferon-gamma (IFN-γ) deficiency called the role of the Th1 lineage in autoimmune
pathogenesis into question (1-3). The recent discovery of another subset of helper T cells,
termed Th17, breaks the long-accepted paradigms about T cell subsets in autoimmunity and also
promises to resolve the contradictory evidence regarding the role of Th1 cells. Identified
originally by their expression of the pro-inflammatory cytokine IL-17, Th17 cells have gained
wide acceptance as a distinct subset of CD4+ cells with expression of unique master regulatory
transcription factors retinoic acid receptor-related orphan receptor-gamma-t (RORγt) and RORα
(4, 5). Th17 cells are induced by a number of pro-inflammatory cytokines including IL-1, IL-6,
and IL-23, and in turn these cells secrete anti-microbial peptides and pro-inflammatory cytokines
such as TNF, IL-21, and IL-22.
Given that the populations of Th17 cells and Tregs exist in reciprocal proportions in both healthy
and diseased tissues, (6, 7) elucidating the relationship between these lineages may yield critical
insight into the disruption of immunological homeostasis. However, our current data about the
interaction and relationship between these subsets is incomplete and at times contradictory.
Initially it was believed that these two lineages were mutually exclusive phenotypes that shared a
common progenitor and was determined by the local cytokine milieu. In the presence of TGF-,
commitment of naive CD4+ T cells to the Th17 or Treg lineage depended on the presence or
absence of IL-6, respectively (6). In support of the view that the lineages are exclusive and
develop reciprocally, Tregs antagonize Th17 cell development by suppressing expression of
RORγt (8), while retinoic acid acts as a switch that suppresses Th17 expansion and enhances
Treg proliferation (9).
However, recent studies suggest that the relationship between these two lineages cannot be
characterized as a rigid dichotomy, but rather is more nuanced. In murine models, IL-17
production could be induced independent of IL-6, and naïve Th17 cells could be generated in IL6 KO mice following Treg depletion (10, 11). Initial studies of the differentiation of human
Th17 cells also questioned the role of TGF-β in the commitment to this lineage, but others have
since identified TGF-β as a critical cytokine for the induction or amplification of this lineage in
combination with various cofactors, including IL-6, IL-1, IL-23, and IL-21(5, 12). Data suggest
that TGF- amplifies Th17 expansion by preventing the maturation of the antagonistic Th1
lineage (13) and Tregs may actually facilitate Th17 commitment, through provision of TGF-
and the induction of TGF- from antigen-presenting cells (14). Thus, the cytokines once
considered critical to the development of this lineage as well as the relationship between Th17
cells and Tregs remain incompletely elucidated.
Furthermore, accumulating evidence suggests that cells that are typically considered to be
terminally differentiated Tregs in fact retain considerable phenotypic plasticity and can develop
into cells that exhibit the characteristic Th17 phenotype. In the presence of immune modulator
B7-DC XAb-, dectin-1-, or TLR4-activated dendritic cells, Tregs can be induced to downregulate Foxp3 expression and produce IL-17 instead (15-17). A recent study by Zheng et al
suggests that inducible Tregs (iTregs) may be more resistant to this conversion than natural
Tregs (nTregs), although activating nTregs with IL-2 and TGF- increased their resistance (18).
ROR-γ-t but IL-17- T cells that co-express archetypal Treg cell markers (CD25 and Foxp3) have
been observed in a variety of murine tissues, and Tregs apparently differentiate into IL-17
producers in the murine small intestine in vivo (8, 19). Thus, such phenotype-switching is not
restricted to artificial culture systems, but may occur in response to fluctuations in the local
cytokine milieu in vivo. Conversely, restimulation of murine Th17 cells with IL-6 and TGF-
(in contrast to IL-23) induces a nonpathogenic phenotype that coexpresses IL-17 and IL-10,
arguing that certain subsets within the Th17 lineage may exhibit regulatory rather than proinflammatory functions under certain conditions (20).
In the context of the mechanisms by which stimulated Tregs differentiate into a subpopulation of
IL-17 producing T cells, my project is designed to explore the mechanisms of the reverse
phenotype-switching: namely, the exogenous factors that can induce differentiated Th17 cells
into T cells with a characteristic Treg phenotype (typified by the expression of transcription
regulator Foxp3, and costimulatory factors CD25 and CD127 (21).) In addition to cytokine
expression, I will examine the resulting population for stability/memory and characteristic Treg
function by determining the duration of Treg-marker expression and the population’s capacity to
suppress T effector cells, respectively. Previous related work has led to the identification of
vitamin A metabolite all-trans retinoic acid and rapamyacin as cofactors that inhibit Th17
differentiation and drive Treg expansion; however, these studies examined unfractionated cell
populations or naïve CD4+ precursors rather than fully differentiated Th17 cells (9, 21-22).
Thus, my project will contribute to the elucidation of the phenotype-switching and the degree of
plasticity exhibited by highly-differentiated T cells by focusing on the under-studied mechanisms
by which Th17 cells expressing characteristic pro-inflammatory cytokines can be polarized to
display the Treg phenotype. In the broader context of immunological homeostasis and
autoimmunity, the aim of this project is to explore mechanisms that can drive an imbalance
between T cell populations; more knowledge in this field will be foundational for the design of
highly-targeted and specific therapies aiming to abrogate autoinflammation by inducing immune
regulation in diseased tissues.
Main Objective
To determine the extent to which exogenous all-trans retinoic acid induces helper T cells
committed to the Th17 lineage to lose the characteristic Th17 phenotype and attain a Treg
phenotype (Aim 1); To assess the memory/stability of this population by examining the duration
of the Treg phenotype (Aim 2); To characterize the extent to which this population exhibits
characteristic Treg function by determining its capacity to suppress T effector cells (Aim 3)
Methodology
Aim 1: Inducing Phenotype-Switching
Cell isolation, polarization, and enrichment: Buffy coats of whole blood will be subjected to
density-gradient centrifugation as prescribed by the standard procedure used for previous
experiments in the Mellins lab. Adapting the already-optimized procedure outlined in “Rapid ex
vivo isolation and long-term culture of human Th17 cells” by Streeck et al. (23), CD4+ T cells
will be isolated by negative selection with magnetic bead (MACS) sorting and stimulated under
non-lineage specific polarizing conditions for T cells (with a combination of PMA, ionomycin,
costimulatory factors CD49d and CD28.) This population will then be enriched for IL-17
producing T cells by conjugating anti-IL-17 antibodies to avidin to form a highly specific capture
complex as detailed by Streeck et al.; using this procedure, two rounds of positive selection have
been shown to yield a purity of over 95% (23). Purity will be quantified by intracellular staining
for IL-17 and FACS.
Characterizing the effects of Retinoic Acid: Th17 cells will be cultured with exogenous retinoic
acid and incubated overnight; the following morning cells will be transferred to fresh media and
their phenotype assessed through flow cytometry. Expression of characteristic Treg markers (low
CD127, high CD25) will be contrasted with expression of Th17-specific IL-17, CCR6, and
CCR4. Quantitative PCR (qtPCR) will be performed in addition to assess the expression of
transcriptional regulators of Foxp3 relative to ROR--t.
Aim 2: Characterizing Stability
Long-term cultures: The aforementioned population of enriched Th17 cells will be expanded in
vitro for use in long-term cultures using the procedures outlined by Streeck et al. (23). Briefly,
Th17 cells will be maintained in R10 medium in the presence of IL-2; these cells will be
transferred to fresh media three times each week. Cultures will initially be stimulated on the
second day of incubation with irradiated feeders and T cell stimulatory anti-CD3/anti-CD8 and
re-stimulated every 3 weeks.
Characterization of phenotype: Phenotype of Th17 cells incubated with exogenous retinoic acid
will be determined at five distinct time points (6 hours, 24 hours, 2 days, 4 days, 6 days) over a
week-long period. (Cells will be incubated with retinoic acid overnight and then transferred to
fresh media. Time points assessed will be subject to change depending on the results of
preliminary data.) Expression of characteristic Treg markers (low CD127, high CD25) will be
assessed through flow cytometry and contrasted with expression of IL-17, CCR6, and CCR4
(chemokine receptors confirmed as prototypical Th17 markers by Acosta-Rodriguez et al. (24).)
Aim 3: Assessing Function
Suppression Assays: These assays will be modeled on the suppression assays described in
studies examining phenotype-switching from Tregs to Th17 cells (21, 24) in addition to similar
assays previously performed in the lab. In brief, the Th17 cell population will be co-cultured
with CFSE-labeled CD4+CD25- T effectors (Teff) (isolated and enriched through a procedure
similar to the one detailed above) 48 hours after incubation with retinoic acid. The expansion or
decline of the Teff cell population will be quantified through flow cytometry (with intracellular
staining with CD25 and IL-2 to differentiate between the Treg and Teff populations.)
Experimental Timeline
6/2008-8/2009: Self-directed reading on Th17 cells with particular emphasis on their interactions
with Tregs. I plan to continue to read two experimental articles per week from established
journals in the field to keep myself updated on the most recent work in the field and gain
background knowledge in experimental design.
6/22-7/3 (two weeks)
I will spend the first two weeks refamiliarizing myself with cell-isolation and –polarization
procedures by generating and enriching populations of Th17 cells from whole blood. I will
check these populations for purity with a goal of at least 85% of all cells positive for IL-17. The
populations isolated during these weeks will form the basis for my first long-term cultures,
which I will maintain and check for cell vitality at least twice a week. I will run a preliminary
experiment assessing the effects of incubation of these cells with retinoic acid following the third
isolation.
7/6-7/17 (two weeks)
This time will be spent characterizing the effects of incubation with retinoic acid. I will repeat
the procedures outlined for this above (see Aim 1) at least three times during this period, using a
combination of qt-PCR and flow cytometry to assess the phenotype of these cells. In addition, I
will prepare at least two buffy coats to expand my long-term cultures. Previously-established
cultures will be maintained.
7/20-8/7 (three weeks)
Stability assays as described above will be performed during this period. Each Monday and the
second Wednesday of this period I will start a new week-long assay from my previouslyestablished cultures for a total of four sets of data. Depending on the vitality of previous
cultures, one or more buffy coats will be processed for the establishment of new long-term
cultures of Th17 cells; existing long-term cultures will be maintained.
8/7-8/29 (three weeks)
Suppression assays will be performed the first two Mondays and the first Wednesday, leaving
the final week open for another suppression assay or a repetition of stability or phenotypical
assays. Long-term cultures will be maintained for the first two weeks, and the third week will be
primarily devoted to a comprehensive assessment of data as a whole. I plan to present an
overview of my findings to the lab late in the last week during lab meeting.
Resources
This project will be carried out under the supervision of principal investigator Professor
Elizabeth Mellins, and postdoctoral fellow Dr. Claudia Macaubas. My internship this past
summer has familiarized me with the workings and dynamics of the Mellins lab, which will
provide me with the necessary equipment and reagents to carry out my project.
Preparation
Although I have not begun any wet-lab work on this particular project, my internship this past
summer in Betsy Mellins’ lab in the Stanford Medical School introduced me to lab techniques
(in particular, immunologic-based assays and equipment including Western-blotting, cell-sorting
and –staining, PCR, and flow cytometry) that will be required for this project. I worked with a
different cell type this past summer but feel comfortable adapting the techniques I practiced with
monocytes to T cells and gained experience in trouble-shooting experiments that initially result
in unsatisfactory isolations and low yields. Additionally, I have been reading extensively on the
subject of Th17 cells since the middle of last summer and currently have a review concerning the
involvement of Th17 cells in rheumatoid arthritis under review for publication in Clinical
Immunology. My familiarity with both the subject-matter and the lab procedures will facilitate
my work this summer.
Budget
$2000 Housing through the Summer Research College (6/16/08-9/05/08)
$1500 19-meals a week meal plan
$1700 Summer Stipend (12 weeks)
Total = $5200
References
1. Takayanagi, H. et al. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling
cross-talk between RANKL and IFN-. Nature 408: 600-605.
2. Huang, W. et al. 2002. Exposure to receptor-activator of NFB ligand renders preosteoclasts resistant to IFN- by inducing terminal differentiation. Arthritis Res Ther 5:R49.
3. Sato, K. et al. 2006. Th17 functions as an osteoclastogenic helper T cell subset that links
T cell activation and bone destruction. The Journal of Experimental Medicine 203:26732682.
4. Yang, X. O. et al. 2008. T Helper 17 Lineage Differentiation Is Programmed by Orphan
Nuclear Receptors ROR and ROR. Immunity 28:29-39.
5. Mangan, P. et al. 2006. Transforming growth factor- induces development of the Th17
lineage. Nature. 441:231-234.
6. Bettelli, E. et al. 2006. Reciprocal developmental pathways for the generation of pathogenic
effector Th17 and regulatory T cells. Nature. advanced online publication: 235-238.
7. Mucida, D. et al. 2007. Reciprocal Th17 and Regulatory T Cell Differentiation Mediated by
Retinoic Acid. Science. 317 (5835): 256-260.
8. Zhou, L. et al. 2008. TGF--induced Foxp3 inhibits Th17 cell differentiation by antagonizing
RORt function. Nature 453:236-240.
9. Xiao, S. et al. 2008. Retinoic Acid Increases Foxp3+ Regulatory T Cells and Inhibits
Development of Th17 Cells by Enhancing TGF--Driven Smad3 Signaling and Inhibiting IL-6
and IL-23 Receptor Expression. J Immunol 181:2277-2284.
10. Korn, T. et al. 2007. IL-21 initiates an alternative pathway to induce proinflammatory Th17
cells. Nature advanced online publication:484-487.
11. Kimura, A. et al. 2007. IL-6-dependent and -independent pathways in the development of
interleukin 17-producing T helper cells. Proceedings of the National Academy of Sciences
104:12099-12104.
12. Manel, N. et al. 2008. The differentiation of human Th-17 cells requires transforming growth
factor- and induction of the nuclear receptor RORt. Nat Immunol 9:641-649.
13. Annunziato, F. et al. 2008. The phenotype of human Th17 cells and their precursors, the
cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int.
Immunol. 20:1361-1368.
14. Xu, L. et al. 2007. Cutting Edge: Regulatory T Cells Induce CD4+CD25-Foxp3- T Cells or
Are Self-Induced to Become Th17 Cells in the Absence of Exogenous TGF-beta. J Immunol
178:6725-6729.
15. Xu, L. et al. 2007. Cutting Edge: Regulatory T Cells Induce CD4+CD25-Foxp3- T Cells or
Are Self-Induced to Become Th17 Cells in the Absence of Exogenous TGF-beta. J Immunol
178:6725-6729.
16. Radhakrishnan, S. et al. 2008. Reprogrammed FoxP3+ T Regulatory Cells Become IL-17+
Antigen-Specific Autoimmune Effectors In Vitro and In Vivo. J Immunol 181:3137-3147.
17. Fabiola, O. et al. 2008. DC activated <I>via</I> dectin-1 convert Treg into IL-17 producers.
European Journal of Immunology 38:3274-3281.
18. Zheng, et al. 2008. Cutting Edge: Foxp3+CD4+CD25+ Regulatory T Cells Induced by IL-2
and TGF-{beta} Are Resistant to Th17 Conversion by IL-6. J Immunol 180:7112-7116.
19. Lochner, M. et al. 2008. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL10+ Foxp3+ RORt+ T cells. J. Exp. Med.:jem.20080034.
20. McGeachy, M. J. et al. 2007. TGF-[beta] and IL-6 drive the production of IL-17 and IL-10
by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol 8:1390-1397.
21. Liu, W. et al. 2006. CD127 expression inversely correlates with FoxP3 and suppressive
function of human CD4+ T reg cells. The Journal of Experimental Medicine. 203 (7): 170111.
21. Elias, K. M. et al. 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3
expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111:1013-1020.
22. Kopf, H. et al. 2007. Rapamycin inhibits differentiation of Th17 cells and promotes
generation of FoxP3+ T regulatory cells. Int Immunopharmacol 7 (13): 1819-24.
23. Streeck, H. et al. 2008. Rapid ex vivo isolation and long-term culture of human Th17
cells. J Immunol Methods 333: 115-125.
24. Acosta-Rodriguez, E. 2007. Surface phenotype and antigenic specificity of human
interleukin 17-producing T helper memory cells. Nature Immunology 8: 639-646.