Open Access version via Utrecht University Repository
advertisement

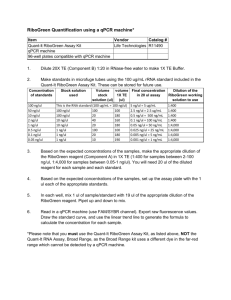
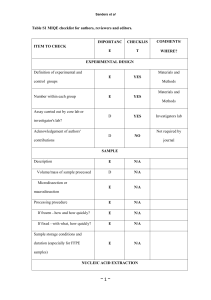
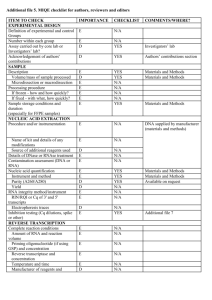
Development of a qPCR assay for diagnosis of Mycobacterium avium subsp. Paratuberculosis (MAP) infection in Deer Onderzoekstageverslag Marielle Buijs Studentnummer: 0247839 23 april 2009 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Table of Contents 1. Abstract ......................................................................................................2 2. Introduction ................................................................................................3 3. Materials & Methods ....................................................................................10 3.1. Herds ............................................................................................10 3.2. Method of sampling 3.3. Sample processing .........................................................................11 3.4. Fecal DNA extraction ...................................................................11 3.5. Lymph node tissue DNA extraction .............................................12 3.6. PCR (Endpoint) ..............................................................................13 3.7. PCR (Quantitative) .......................................................................13 ...................................................................10 4. Results...........................................................................................................17 5. Discussion ................................................................................................26 6. Conclusion ..................................................................................................31 7. References ................................................................................................33 1 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 1.Abstract Mycobacterium avium subspecies paratuberculosis (MAP) is the cause of Johne’s disease, a chronic infectious enteritis, in ruminants. Johne’s disease causes significant losses due to clinical disease in deer <15 months of age. The most common route of infection is ingestion via contaminated pasture, food and water contaminated by MAP infected faeces. For management of JD, early identification of heavy shedders from a faecal sample would be a useful tool in preventing further pasture contamination and spread of infection. Several diagnostic tests are available, including faecal culture and an serological immunoassay, which are limited by lack of sensitivity, specificity and long incubation times. Polymerase Chain Reaction (PCR) assays have been applied for detection of MAP, these PCR assays have shown to be sensitive and specific for the detection of MAP and have reduced time for detection to 2-3 days. The central objective of this study was to develop a fast, robust and reproducible qPCR assay for MAP in faecal material and tissue samples of deer. Two different MAP sequence targets were compared (F57 and IS900) and two different detection chemistries were compared (SYBR Green vs. TaqMan). A comparison with an established immunological bioassay, an ELISA (Paralisa) has been made. A significant correlation between the Cervine matched blood, (Paralisa Johnin results), and faecal samples(qPCR CT values) is verified, p < 0.001. Finally the effect of lab contamination was evaluated. This study revealed that it is possible to do a qPCR assay to detect MAP. 2 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 2. Introduction Paratuberculosis, or Johne’s disease (JD), is a chronic infectious enteritis of domestic and wild ruminants (15,23,39) which has a significant and detrimental impact on primarily agricultural economies such as that of New Zealand (20). In addition, a potential link between paratuberculosis and the human condition known as Crohn’s disease has been hypothesised and, while evidence remains controversial, paratuberculosis is considered as a public health concern (5, 37). Paratuberculosis is caused by Mycobacterium avium subspecies paratuberculosis (MAP), a hardy, slow-growing, gram-positive, and acid-fast bacterium (16, 45). Despite 99% genetic identity, MAP can be distinguished phenotypically from the closely related M. avium subspecies avium and M. avium subspecies sylvaticum by its dependence on mycobactin for growth in culture (5) and genotypically by the presence of multiple copies of a characteristic insertion element, IS900 (6, 13). Ingested MAP bacteria enter the intestinal wall through the small intestinal mucosa, primarily in the region of the ileum, via M cells covering the Peyer’s patches (45). MAP are phagocytosed by resident subepithelial macrophages, where they are able to inhibit phagolysosomal fusion, resulting in resistance to intracellular degradation (45). While the bacteria are in the mucosal tissue and submucosal macrophages, there is little or no detectable immunological reaction to the infection. The delayed humoral immune response is one reason for the poor sensitivity of serological tests for MAP. Eventually the infected macrophages migrate into local lymphatics, spreading the infection to regional lymph nodes (45). In the regional lymph nodes, the organisms are capable of stimulating inflammatory and immunological responses. There are several characteristics that animals affected by Johne’s disease can display sub-clinically and clinically. In the initial stages of disease in deer, the animal fails to thrive, stops growing and the weight and condition both decrease (25). In spring the affected animal only partially moult. A large part of the animals affected by the disease also exhibit diarrhea. The total time span of the clinical stage of the disease is a couple of weeks. There is a variation between younger and older animals. Animals which are infected at a younger age seem to die earlier of the disease (23, 25). Johne’s disease causes significant losses due to clinical disease in deer <15 months of age (23, 24). 3 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer In dairy herds, similar clinical symptoms are observed; the sub-clinical form of the disease in cattle results in reduced milk production, with possible impacts on fertility and udder health and premature culling. Eventually infection can lead to the clinical form that manifests as progressive weight loss, lower slaughter value, chronic diarrhea, emaciation, debilitation and eventual death (45). Paratuberculosis in sheep and cattle differs to deer in the fact that they usually show clinical signs by the age of 3-5 years (26). Macroscopically, granulomatous lesions are most often identified at post mortem within the mesenteric lymph nodes in the region of the ileum and the jejunum and occasionally within the ileocaecal lymph nodes also. Enlarged lymph nodes may exhibit caseous lesions also. The lacteal ducts from the jejunum to the adjacent lymph nodes become inflamed and often appear opaque in appearance (lymphangitis). In sheep and cattle there is also thickening of the intestinal wall (26, 45). The most common route of infection is ingestion via contaminated pasture, food and water contaminated by MAP infected faeces (26). To prevent infection, it is recommended that sheep or cattle should not be grazed on the deer farm, unless they are known to be ‘negative animals’. Other measures include keeping a closed herd and the use of artificial insemination. Better control of this disease requires a greater knowledge of the biology of MAP, its epidemiology, interspecies transmission, strain diversity and contribution of wildlife reservoirs of disease (44). Genetic resistance to mycobacterial infection in deer has been described with an estimated heritability of resistance to m. bovis reported to be 0.48 in red deer; in the future the use of resistant breeding stags may complement existing Tb control programmes (27). Johne’s disease in New Zealand was first reported in 1979 (10, 15). More recently it has became clear that Johne’s disease is much more widespread in NZ than initially thought (24). With a modern day national herd approaching 1.3 million deer in New Zealand, spread over about 3,051 Farms, and with regard to the financial impact that the disease has (28), it is essential to have robust, reliable, sensitive and specific diagnostic detection methods. When it comes to (post mortem) JD diagnosis, culture of the organism from tissue samples taken at necropsy is still widely considered as the ‘gold standard’ reference test as, even before faecal shedding occurs or a humoral immune response has had a chance to develop, highly sensitive, radiometric culture techniques (BACTEC) are capable of detecting growth of MAP in multiple organs including the intestinal mucosa and submucosa and 4 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer regional lymph nodes (45, 47). BACTEC culture provides a growth index of viable organisms and the number of viable organisms (33). However, as MAP grows extremely slowly in culture, it can still require a period ranging from weeks to months to obtain results from culture and, other than cost, this is considered to be the primary limitation of bacterial culturing as a diagnostic method. Ante mortem tests include faecal culture, serological immunoassay to detect reactivity to mycobacterial antigens by ELISA and, increasingly, nucleic acid amplification based approaches to detect mycobacterial DNA in samples. A derivative of ELISA, the Paralisa test provides a customized ELISA, based on the antigen reactivity of the IgG1 isotype, for the serodiagnosis of Johne’s disease in deer and has a reported specificity of 99.5% and has a sensitivity of 84% with Purified Protein Derivative (PPDj) antigen, but the test is still 75% less sensitive for animals that are positive in culture and have no detectable pathology (14). Molecular analysis of nucleic acids using Polymerase Chain Reaction (PCR) assays have been applied for detection and of MAP in faecal samples, milk and tissue (8, 19, 39). These PCR assays have shown to be sensitive and specific for the detection of MAP and have reduced the time for detection to 2-3 days compared to other diagnostic assays, including enzyme-linked immunosorbent assays (ELISA) and faecal culture, which are limited by lack of sensitivity, specificity and long incubation times (8, 19). As faecal contamination is considered one of the primary routes of infection, it means that when the animal is a high shedder, it can take 2-4 months before the actual cause of JD is determined and by that time the animal is responsible for significant faecal spreading of MAP on the pasture giving the opportunity to other animals in the herd to become infected. A faster method of diagnosis is necessary to prevent spreading of the disease (19). The IS900 sequence is the most commonly targeted sequence for molecular confirmation of MAP, despite some concerns that the sequence may not be 100% unique to MAP (8, 11). Due to homologous, IS900-like elements, in related mycobacterial species, cross-reactions can potentially result false-positive results (9, 12) in rare cases. The IS900 sequence is 1,451 base pairs in length and occurs in 14 to 20 copies per genome (6, 8, 13). For greater specificity of PCR of MAP, other genes, which have a lower copy number in the MAP genome, and thus a potentially lower analytical sensitivity, than IS900, such as F57 (32), hspX (2), ISMav2 (42) and ISMap02 (41) have been suggested (30, 31). However, the reliability of these methods is dependent upon overcoming inherent shortcomings that nucleic acid-based methods have, namely cell lysis efficiency, DNA absorption to surfaces and PCR inhibition from co-extracted compounds present in environmental samples (8). Improved 5 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer DNA extraction protocols, that effectively remove inhibitors will likely result in a increased sensitivity for PCR based methods (39). Due of the impact of the disease on animal health and welfare, production losses in deer farmed for venison and the looming threat of a zoonotic association with Crohn’s disease, a rapid, cost-effective, and automated diagnosis of this pathogen can still be regarded as a high priority task (39). PCR based tests are an attractive prospect in terms of speed and cost but, like any diagnostic assay, must be carefully controlled for both false-positive and false-negative results. For management of JD, early identification of heavy shedders from a faecal sample would be a useful tool in preventing further pasture contamination and spread of infection; such a quantitative assay may be accomplished with a quantitative PCR (qPCR) approach. A qPCR assay should fulfill criteria such as a high detection probability in the tissue to be examined, straightforward sample preparation and DNA extraction, high specificity, robustness, reproducibility and working with a user-friendly protocol (4). While several articles have suggested that PCR analysis cannot compete with the sensitivity of faecal culture of MAP (43, 46), others report that detection of MAP in ovine faeces by direct quantitative PCR has similar of greater sensitivity compared to radiometric culture (19). As methodologies for extracting high-quality, inhibitor-free DNA from faecal material improve, sensitivity and reproducibility of faecal PCR diagnostic tests will likely improve. Mechanism of qPCR compared to conventional (endpoint) PCR: An endpoint PCR assay examines by agarose gel electrophoresis the final amount of DNA amplicon produced after a defined number of cycles and the result is determined by the presence or absence of a DNA band of the expected size. Endpoint PCR does not provide information about quantity of starting material in the reaction and returns a ‘yes/no’ result with regard to presence of target sequence. A qPCR assay, however, measures the amount of DNA target as it accumulates during a PCR reaction and the cycle number at which product begins to be detected is directly proportional to the starting quantity of target template present in the initial sample. The cycle number at which the target begins to be detected is called the Threshold Cycle (Ct) and is determined early in the exponential phase of the reaction (Figure 1). Accumulation of product is detected through an increase in a fluorescent reporter and is monitored throughout the reaction; a sample containing a higher starting amount of DNA target gives rise to an earlier increase in detectable fluorescence. Thus the Ct value is inversely proportional to the starting copy number of the target and an increase in sample concentration gives a linear increase in fluorescence over a very broad range (3, 4). Detection 6 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer chemistries differ between manufacturers but essentially can be divided into non-specific, DNA binding flourophors (SYBR Green) and dual-labeled hybridisation probes (TaqMan). Figure 1. CT value is determined when the Fluorescence signal rises above background noises, during the exponential exponential growth phase. Non-specific DNA-binding fluorophores (SYBR Green I). The setup of a SYBR Green assay is straightforward and it’s advantage over probe based assays is cost – SYBR assays do not require the commercial synthesis of a dual-labelled hybridisation probe as the flourescent dye is incorporated directly in the reaction mix. The main disadvantage of SYBR assays, however, is that their lower initial cost may sacrifice specificity. SYBR green binds to the minor-groove of double stranded DNA molecules and fluoresces under excitation, thus overall fluorescence increases proportionally to the double stranded DNA concentration; when free in solution the dye does not exhibit fluorescence (3). The specificity of SYBR assays is entirely dependent on the specificity of the primers used as all double stranded DNA generated throughout the assay will bind the dye; any non-specific amplification products, including primer-dimers, will result in false signal and false positive data. For this reason, dissociation plots are analysed after every run to check for non-specific priming or multiple products of differing melting points. The identification of multiple peaks in a dissociation plot, however, cannot rescue the data and serves only to alert the user to an imperfect assay requiring a repeat. 7 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Sequence-specific probes (TaqMan) used in qPCR: TaqMan assays use three sequence-specific oligonucleotides, two conventional PCR primers which flank the target sequence and a labeled hybridisation probe which binds internally to the primers. The probe is labeled with a 5΄ reporter dye and a 3΄ quencher dye and fluorescence is generated during the PCR extension step by strand displacement. This step cleaves the annealed probe through the 5΄ nuclease activity of the Taq polymerase. The fluorophore (R) becomes separated from the quencher (Q) and a fluorescent signal is emitted upon excitation (Figure 2). Fluorescence is directly proportional to the amount of product formed. A multiplex qPCR can be designed using probes labeled with different fluorophores which fluoresce at different wavelengths and can be detected independently by appropriate hardware. Multiplexing is not possible in this fashion with SYBR green assays. Figure 2: a denature: shows primer and probe fluorophore(R) and quencher (Q) are attached to the probe) in solution Anneal: the primers and the probe anneal to the single DNA strand. Extend: As the Taq polymerase extends the chain, the fluorophore becomes separated and emits fluorescence. 8 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer The primary aim of this study was to develop a fast, robust and reproducible qPCR assay for MAP in faecal material and tissue samples. Two different MAP sequence targets were compared (F57 and IS900) and two different detection chemistries were compared (SYBR Green vs. TaqMan) A secondary aim was to compare this qPCR assay to an established immunological bioassay, an ELISA (Paralisa), and investigate how these results might relate to each other. Finally we attempted to quantify the effect of laboratory contamination on PCR results. 9 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 3. Materials & Methods 3.1 Herds MAP isolates were collected from 84 deer, originating from 9 different herds in New Zealand. Herds were located in the following regions: Wanaka, Mid Canterbury, Geraldine, South Canterbury, Balclutha and Fairlie on the South Island and Fielding and Rotarua on the North Island as illustrated in Figure 3. The herds were selected on the basis of clinical diseased animals, with common signs of weight loss and diarrhea and randomly selected clinically healthy animals. From the animals that were not to be culled, we aseptically collected blood and faeces. From the animals that were to be culled, we collected blood before the animals were slaughtered and faeces and tissue lymph nodes after they had been bled. Figure 3: Red spots indicate the regions from which the deer used in this study originated. 3.2 Method of Sampling Blood sampling was performed by using a new needle and a new vacutainer, containing EDTA anti-coagulant for each animal. Blood was taken from the jugular vein by venepuncture, without using tranquillization. Faecal sampling was performed by anal stimulation of the deer using a new latex glove each time and putting it in an aseptic plastic container. Faecal sampling at the slaughter plant was performed by making a cut into the deer jejunum aseptically, after taking tissue from the lymph nodes, directly from the carcass by using sterile instruments and new gloves and sterile plastic cups each time. Tissue sampling was performed aseptically by using new, sterile instruments and new gloves for each deer. 10 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 3.3 Sample Processing Blood samples were processed directly, the tissue samples were stored frozen at -20ºC before using them. Faecal samples were stored at 4ºC prior to processing. 3.4 Fecal DNA Extraction Faecal DNA extraction was performed using the ZYMO Research Faecal DNA Kit TM (ZYMO Research Corp., USA) using a modified protocol previously developed at the DRL. Briefly, for each sample 2-3 faecal pellets (3g) (or an equivalent volume of liquifactious faeces) was transferred into a sterile 50 mL polypropylene centrifuge tube which had been pre-prepared by putting in approximately 5 mL of 4 mm sterile, glass beads and 20 mL of sterile water. Each sample was vortexed vigorously for 2 minutes to completely homogenize the faecal sample and a wide-bore pipette was used to transfer 1.2 mL of the homogenate to a sterile, 2 ml, screwtop microcentrifuge tube containing 0.5 mL of 0.1 mm sterile, zirconia beads. Samples were centrifuged at 13k rpm (13k g) for 2 minutes to pellet the insoluble component of the sample onto the beads and supernatant discarded. This resulted in a 2-3 mm thick band of fecal material overlying the beads. 750 μL of Lysis Buffer was added to the tube and beads and sample thoroughly resuspended. The tubes were tightly capped and placed in a boiling water bath for 5 minutes; after this the samples were transferred to a Disruptor Genie apparatus (Scientific Industries Inc., USA) to bead-beat samples for 15 minutes. This compound DNA extraction approach included chemical, thermal and mechanical steps to ensure maximal disruption of the mycobacterial cell wall and efficient lysis of mycobacteria. The samples were centrifuged at room temperature, 13k rpm for 1 minute and 400 μL of the supernatant transferred to a Zymo-SpinTM IV Spin filter placed in a 2 mL collection tube to remove any remaining fine, particulate matter. The samples were centrifuged at 7k × g for 1 minute, the filter discarded and 1.2 mL of Faecal DNA Binding Buffer was added to the filtrate from each of the samples in the collection tube. The samples were mixed by pipetting and 800 μL of the sample was added to the ZymoTM IIC Column, to bind the DNA, in a 2 mL collection tube. The samples were centrifuged at 10k × g for 1 minute and the remaining 800 μL of the sample was added to the DNA binding column and centrifugation step repeated. The binding column was washed with 200 μL of DNA Pre-Wash Buffer and again with 500 11 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer μL of Faecal DNA Wash Buffer. The DNA binding column was finally placed in a fresh 1.5 mL microcentrifuge tube and sample eluted with 100 μL of Elution Buffer. In a final cleanup step, the samples were run through a Zymo-SpinTM IV-HRC column and the recovered DNA sample quantified by UV spectroscopy and diluted to 50 ng/μL before performing PCR or qPCR. 3.5 Lymph Node Tissue DNA Extraction Cellular DNA was extracted from lymph node tissues using a standard Proteinase K digestion and organic solvent extraction method (1, 35). In order to process as much of the lymph node tissue as possible and reduce sampling error, the greater part of the tissue sample was initially homogenised. Briefly, after thawing each lymph node sample was placed inside a Class II Biosafety cabinet and rinsed free of external, surface contaminants with sterile water. The lymph node was finely sliced into pieces approaching 5mm before being transferred into a sterile, 15 mL tube filled with 4 mL of TE (Tris EDTA; 10mM Tris.HCl pH 7.9, 1mM EDTA) buffer and homogenised vigorously using an Omni-TipTM Disposable Generator Probes (Omni International, USA). Following homogenisation, 400 μL of the tissue homogenate was removed to a fresh microcentrifuge tube and centrifuged at 13k rpm for 15 minutes. The supernatant was discarded and 400 µL DNA Extraction Buffer (10 mM Tris.HCl pH 7.9, 25 mM EDTA, 100 mM NaCl, 0.5% SDS) was added to each pellet. Twenty microlitres of Lysozyme (10 mg/mL; Sigma Chemical Co., USA) was added each tissue suspension and the samples incubated at 37 ºC for 1 hour (1). Following lysozyme digestion, 10 μL Proteinase K (10 mg/ml; Sigma Chemical Co., USA) was added and the samples incubated at 57ºC for 2 hours to overnight. Five hundred microlitres of Phenol:Cloroform:IsoamylAlcohol (25:24:1, Sigma Chemical Co., USA) was added to each digested sample, mixed vigorously to form an emulsion and centrifuged at room temperature at 13k rpm for 10 minutes to separate aqueous and organic phases. Approximately 300 µL of the top, aqueous layer containing the nucleic acids was carefully removed to a fresh microcentrifuge tube. The DNA sample was made up to 500 µL with TE buffer and NaCl added to a final concentration of 0.2 M. Ethanol precipitation of the DNA was effected by adding 1 mL of absolute ethanol to each sample and centrifuging at 13k rpm for 15 minutes to pellet the DNA. Finally, the pellet was washed free of salts with a final wash of 70% ethanol and air dried. The resulting DNA pellet was redissolved in 500 μL TE 12 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer buffer and the DNA samples quantified by UV absorption spectroscopy. Each tissue sample was diluted to a final concentration of 40ng/µL. 3.6 PCR (Endpoint) To confirm that high quality DNA, free from coextracted PCR inhibitors had been recovered from faecal and lymph node samples, conventional PCR was performed on known positive samples. The target sequence IS900 was amplified using primers P90 (5΄GTTCGGGGCCGTCGCTTAGG- 3΄) and P91 (5΄ -GAGGTCGATCGCCCACGTGA- 3΄) to produce a 400 bp fragment (29, 36). Endpoint PCR reactions were carried in a 30 µL final volume and were prepared as listed in Table 1, below. Component Final Concentration * 10 PCR Amplification Buffer P90/P91 Primer Mix (20 µM) HotMaster Taq DNA Polymerase* dNTPs (10mM) Template DNA Sterile H2O * 1 0.2 µM 1.5U/reaction 0.2mM 500 ng To final volume 5 Prime Inc., USA Table 1. Components of endpoint PCR for IS900 PCR reactions were cycled in a PCR thermocycler (MJ Research Dyad DNA Engine) for 30 cycles which consisted of 30 seconds at 95ºC for template denaturation, 30 seconds at 58ºC for primer annealing and 30 seconds at 65ºC for polymerase extension. PCR products were analysed by gel electrophoresis on a 1% agarose gel prepared with Tris Borate EDTA Buffer (89 mM Tris, 89 mM Boric Acid, 2 mM EDTA), stained with ethidium bromide and visualised under UV light. 3.7 PCR (Quantitative) qPCR amplifications were performed on an ABI 7500 FAST Sequence Detection System (Applied Biosystems, USA). The quantitative polymerase chain reaction was used to measure the relative concentration of MAP DNA in a sample. Preliminary experiments was based on the IS900 target sequence, generally regarded to be specific for MAP. In later 13 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer experiments, the MAP specific sequence F57 was also included. While an assay was designed to amplify the cervine GAPDH gene to function as an internal amplification control, this assay was not investigated due to time restraints and is not discussed here. To confirm the functionality of the test we used in each qPCR assay a sample from a low shedder and a high shedder. The qPCR was performed on the DNA isolated from faecal samples and lymph node tissue, after diluting the concentrations of the DNA down to 50ng/μL for faecal DNA and 40ng/μL for tissue DNA. Primers for qPCR were designed using Primer Express Version 2.0 software (Applied Biosystems, USA) to suit the universal amplification conditions required for qPCR assays. Details of the primers and hybridisation probe sequences are shown in Table 2, below. Sequence (5΄ - 3΄) Assay ID IS900 SYBR IS900 TaqMan Fwd CCGTCGCTTAGGCTTCGA Rev ATCCAACACAGCAACCACATGA Fwd AACGCGCCTTCGACTACAAC Rev CGGGAGTTTGGTAGCCAGTAAG Product Size (bp) 66 Probe FAM-AAGAGCCGTGCCGCGCTGATC-BHQ1 F57 TaqMan Fwd GGCAGCTCCAGATCGTCATTC Rev GCGGTCCAGTTCGCTGTCAT 98 Probe FAM-ATGAATCGGCAGCTACGAGCACGC-BHQ1 Fwd GAPDH TaqMan Rev GGAAAGGCCATCACCATCTTC GGACTCCACCACGTACTCA 84 Probe JOE-CAGGAGCGAGATCCCGCCAACAT-BHQ1 Table 2. Primer information of qPCR Assays A 2 SYBR green master mix was used for the initial set of qPCR reactions (Absolute TM QPCR SYBR Green Low ROX mix, Abgene UK) which included Thermo-Start DNA Polymerase, a proprietary reaction buffer including MgCl2, dNTP’s, SYBR Green I and a ROX passive reference dye. Individual SYBR qPCR reactions were typically carried out in a total volume of 20 µL which consisted of 10 µL Absolute QPCR SYBR Green Mix, 2 µL of DNA template (80 ng and 100 ng in total for tissue and faecal DNA preparations, respectively) and 100 nM of primers appropriate for the assay. For multiple reactions, a mastermix was routinely prepared which included all the reaction components except the DNA template as this allowed for greater pipetting accuracy. The mastermix was prepared and added in 18 µL aliquots to individual wells of a 96 well PCR plate (MicroAmp Optical 14 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 96-well Reaction Plates, Applied Biosystems, USA) and subsequently 2 ul of DNA template was added. Plates were sealed with an optically clear adhesive film (Applied Biosystems, USA) and centifuged briefly to gather all of the reaction mix at the bottom of the wells. All qPCR reactions were performed in duplicate with a thermal cycling programme of 1 cycle at 95º for 15 minutes followed by 40 cycles of 95ºC for 15 seconds and 60º for 1 minute and finally 1 cycle of 95ºC for 15 seconds, 60ºC for 1 minute and 95º for 15 seconds for dissociation curve analysis. Dissociation curves were routinely generated after SYBR assays to verify the size and specificity of the products produced. SYBR reaction components are summarised in Table 3. A 2 TaqMan master mix was used for the probe based qPCR reactions (Absolute TM QPCR Low ROX Mix, ABgene, UK). This mix included essentially the same components as the SYBR mix described above but without the SYBR Green dye. In additon to forward and reverse primers, TaqMan assays incorporate a hybridisation probe – probe and primer sequences are listed in Table 2. The IS900 and F57 probes were each labeled with a 5΄ FAM reporter with maximum absorbance at 494 nm, the GAPDH probe was labeled with the JOE dye with maximum absorbance at 520 nm. All three TaqMan probes were 3΄ labelled with a Black Hole Quencher (BHQ) flourophore (Sigma-Aldrich Co., USA) with maximum absorbance at 534 nm. TaqMan assays were prepared essentially as described for SYBR assays above, with the exception that primer concentrations of 600 nM were used and hybridisation probe was added at 200 nM. Template DNA concentrations were as for SYBR assays. Reaction conditions remained the same although no dissociation curve step was necessary. Reaction Mixes used qPCR-SYBR. 2 Absolute TM qPCR SYBR Green Low ROX Mix 10 μM Forward / Reverse Primer Mix Sterile H2O Template DNA Total Volume Volume Final concentration 10.0 μL 1 0.2 µL 7.8 µL 2.0 μL 20 μL 100 nM 80 to 100 ng 15 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer qPCR-TaqMan-IS900 Volume 2 AbsoluteTM qPCR Low ROX Mix 20 μM IS900 Primer Mix 5μM IS900 Probe Sterile H2O DNA Total volume 10.0 μL 0.6 µL 0.8 μL 6.6 μL 2.0 μL 20 μL Final concentration 1 600 nM 200 nM 80 to 100 ng qPCR-TaqMan-IS900 or F57 + GAPDH (Multiplex) Volume 2 AbsoluteTM qPCR Low ROX Mix 10 μM Primer Mix (IS900 or F57) 10 μM Primer Mix (GAPDH) 5μM Probe (IS900 or F57) 5μM Probe (GAPDH) Sterile H2O DNA Total volume 10.0 μL 0.2 µL 0.2 µL 0.8 μL 0.8 μL 5.2 μL 2.0 μL 20 μL Final concentration 1 600 nM 600 nM 200 nM 200 nM 80 to 100 ng Table 3. qPCR reaction mixes used 16 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 4. Results Faecal DNA extraction Endpoint PCR analysis using primers P90 and P91 demonstrated that DNA free of PCR inhibitors had been successfully prepared from faecal material. The presence of a 400 bp PCR product confimed the presence of IS900 DNA in the sample. Figure 4: An example of an agarose gel showing the result obtained with an endpoint PCR. PCR amplicons at 400 bp confirm the presence of IS900 in each of four samples extracted from cervine faeces. Analytical sensitivity of the SYBR IS900 qPCR assay Starting with a known quantity of MAP genomic DNA originating from cultured material (strain 316f), a serial dilution of MAP DNA was introduced in a sample of cervine DNA extracted from a negative faecal sample in order to determine the smallest amount of MAP DNA which could be detected. Six samples were prepared and assayed in duplicate as shown in Figure 5. As expected, increasing dilution of the MAP DNA resulting in successively higher Ct values in the qPCR. The lowest quantity of MAP DNA which could be reliably detected was approximately 10 genome equivalents (50 femtograms of genomic DNA, 17 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer assuming one MAP genome, at 5 Mbp, to equate to 5 fg (34)). In subsequent assays, a negative cut-point of 35 was used which was less than one genome; Ct values >35 were considered negative. Figure 5. SYBR IS900 qPCR results arising from a faecal sample of a negative animal spiked with decreasing amounts of MAP genomic DNA. 1: 5 ng of MAP DNA (100,000 genome copies); 2: 500 pg MAP DNA (10,000 genome copies); 3: 50 pg MAP DNA (1,000 genome copies); 4: 500 fg MAP DNA (100 genome copies); 5: 50 fg MAP DNA (10 genome copies); 6 (the two lines coming up after “5”): 5 fg MAP DNA (1 genome copy); 7: 0 MAP DNA, negative control reaction. CT values of Individual faecal samples using SYBR IS900 qPCR Of 57 samples available, 53 returned valid results (Figure 6). There were several reasons why some qPCR results were excluded, one sample did not have associated ELISA results, 3 of the samples exhibited poor dissociation curves suggesting nonspecific amplification or primer-dimer artifacts. The 53 samples that were evaluated were classified as highly positive when the CT value was 25. Ct values >25 and 30 were designated as intermediate shedders, Ct values >30 and 35 were considered low shedders Ct values >35 onwards were classified as negative. 18 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Figure 6. IS900 qPCR results from 53 cervine faecal samples showing the range of Ct values obtained. Is there a correlation between qPCR and Paralisa? According to the CT values, each sample was classified a shown below. qPCR Ct Value Classification In Table as 25 High shedder *** >25-30 Intermediate shedder ** >30-35 >35 Low shedder * Negative - The paralisa results were also classified on basis of their results, as shown. Paralisa: Johnin - PPA Classification In Table as 140 and higher 100-140 50-100 below 50 High Intermediate Low Suspect/Negative *** ** * 19 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer According to these classifications, the table below was prepared. Here several similarities are apparent between CT category and Paralisa with Johnin detection antigen result. Paralisa PPA antigen gives less concordance according to this classification, the best similarity is achieved when we take the highest serological result of either Johnin or PPA. Animal ID 37 54 41 38 57 43 51 42 40 34 47 56 50 53 48 45 44 55 36 35 2 20 18 21 29 16 9 27 12 5 23 4 24 11 7 25 19 10 17 26 28 22 14 8 6 3 1 13 31 33 15 30 32 CT category *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** *** ** ** ** ** ** ** ** ** ** ** ** ** ** ** ** * * * * * * * * * * * * * - Highest category of Paralisa *** *** *** *** *** *** *** *** *** *** ** ** ** ** ** ** * * * * *** *** *** ** ** ** ** ** * * ** ** * *** ** ** * * * * * * * * * *** * * - Johnin PPA *** *** *** *** *** *** *** *** *** *** ** ** ** ** ** ** * * * *** *** *** *** ** ** ** * * * *** ** ** * * ** - *** ** ** ** * * ** * * * * *** * ** * * * ** * ** ** * * * * * * * * * * * * *** * * - Table 4. similarities are shown between CT category and Paralisa categories 20 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Raw data Paralisa-Johnin against CT value of matched blood and faecal samples. The trend line gave an R square value of 0.3470. (n=53) 1-0.3470 of the variation is not declared by a linear relationship. The degrees of freedom for entering the t-distribution are N-2. The H0; there is no linear association between the CT value of matched blood and faecal samples ρ = 0. The alternative hypothesis is that ρ ≠ 0. The data for the formula; n=53, r = 0.8435 and r2 = 0.3470 o o According to the t-value is 5.216 5.216 > 3.497 P = 0.001, so P < 0.001, the null hypothesis is rejected. A significant correlation between the Cervine matched blood, (Paralisa Johnin results), and faecal samples(qPCR CT values) is verified, with p < 0.001. Figure 7. Correlation between qPCR generated Ct values and Paralisa Data. CT values acquired by fecal samples compared to lymph node samples The qPCR results obtained from 22 animals for which matched lymph node tissue and faecal samples were available are shown in Figure 8. Generally, the CT value acquired from a lymph node sample was higher than the CT value we acquired from faecal sample from the same animal. The CT values acquired of the lymph nodes lay between 25 and 38; the CT values acquired by the matched faecal samples varied between 14 and 35. 21 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Efficiency of the SYBR qPCR. Green compared to TaqMan assays. Nineteen samples (11 faecal and 9 lymph node samples) were tested with both SYBR Green and TaqMan chemistry. On average the CT values of the TaqMan assay were 2.94 CT lower and the SYBR assay designed for the same target, suggesting that TaqMan assays are on the whole more sensitive than SYBR assays. When it comes to the efficiency of the qPCR, efficiency was both calculated over the same 19 samples in duplicate. Of the SYBR Green assay this was 1.84 and the TaqMan assay this gave a result of 1.91. 22 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer One of the main disadvantages of the SYBR Green qPCR assay was the occurence of unusual amplification plots and/or dissociation curves which suggested that the PCR amplification had been less than optimal in some cases. Often there was no obvious reason for failed PCR and repeated amplification failed to produce useable data. An example of poor SYBR results are shown in Figure 9, below. In Figure 9, Panels A, B and C illustrate successful qPCR, in duplicate, for a sample. Panel A shows a normal SYBR amplification plot, Panel B shows a normal dissociation curve and Panel C a corresponding TaqMan amplification plot. By contrast, Panels D and E show suboptimal amplification from another sample (note unusual form of amplification plot in Panel D and multiple peaks in the dissociation curve shown in Panel E). Finally, Panel F shows the same sample as shown in Panel D amplified successfully with the TaqMan assay. Figure 9. Panel A, Normal SYBR amplification. Panel B, normal SYBR dissociation plot (showing a small double peak as a result of primer-dimer). Panel C, normal TaqMan amplification. Panel D, abnormal SYBR amplification. Panel E, abnormal SYBR dissociation plot. Panel F, correction of poor SYBR assay with use of TaqMan. 23 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer What is the difference between F57 and IS900 in CT value? In a comparison of 18 samples assayed for both F57 and IS900, IS900 was shown to have a consistently lower CT value than F57, with fluorescence detected on average 5.27 cycles earlier (Figure 10). This is not unexpected as IS900 is known to be present in greater copy number within the MAP genome than F57; the fact that the Ct values for IS900 and F57 consistently mirror each other closely confirms that the assay works well. In the figure below is visible how the same samples relate to each other when tested for the two different targets. The consistent difference between the two marker sequences reflects the differing copy number of these genes within the MAP genome. 24 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer Effect of laboratory contamination To study the effect of laboratory contamination , we took some finished PCR product out of a well and prepared a serial dilution series which was analyzed using the TaqMan assay. One μL of PCR product gives a CT value of 2. Contamination with 20 nL (number 2) gives a CT value of 5. Contamination with 0.2 nL gives a CT value between 11 and 12. Contamination with 2 pL gives a CT value of 19. The last one is our negative control animal. A contamination of 2 pL, (according to figure 5, this volume contains 75.000 genome copies, assuming one MAP genome, at 5 Mbp, to equate to 5 fg (34)), made a “low shedder” animal a “high shedder” animal. 25 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 5. Discussion Analytical sensitivity of the SYBR IS900 qPCR assay CT values changed according to different MAP concentrations, in this study, the lowest quantity of MAP DNA which could be reliably detected was approximately 10 genome equivalents (50 femtograms of genomic DNA). In subsequent assays a negative cutpoint of 35 was used, less than one genome (34). A commercial DNA Test Kit is available, VetAlert TM a Tetracore® trademark, this test uses the hspX gene as target sequence and applies TaqMan Chemistry. The positive control in this assay is a non-infectious synthetic template comprising a portion of the target sequence of the hspX gene of MAP. One gene copy is detected at a CT value between 38 and 39. Samples are considered positive when the CT value is ≤38. The positive control, 25,000 genome copies in this system, comes up at a CT value between >24 and <25. The present study shows that with less than 10,000 genome copies a CT value of 21 can be achieved. This difference with the Tetracore kit might be due to the lower copy number of hspX in the MAP genome (2) and due to other differences in the ingredients in the reaction mix, as they may cause a difference in PCR efficiency. This study shows a larger discriminatory range. With a large discriminatory range it is easier to discriminate low, intermediate and high shedders. This may be useful for management of JD. A highly positive animal, shedding high numbers of MAP in the faeces, can be identified and should be culled from the herd with priority. There is also the study of Irenge et al. (18) that uses a Triplex Real Time – PCR. They use IS900, ISMAP02 and f57 –plasmids. Three labeled probes in one tube. With 10 plasmid copies they acquired average CT values of 38.1. The single copy gene F57 was here the limiting factor for the sensitivity of detection of MAP genomic DNA. It doesn’t say what values are acquired by only looking at IS900 fragments, they use three target genes to get more certainty about the product, but the sensitivity seems to be a little bit less, as CT values are slightly higher than in this study. 26 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer figure 1 In this report, as mentioned, less than 5 femtograms of MAP DNA in considered as a negative and not absolutely zero, this is based on the fact that ingested MAP cells can pass through the gut passively for some days after digestion (7, 48). A better positive-negative cut-point for quantity of MAP may need to be validated to distinguish high risk shedders from low risk shedders to provide a good decision making strategy. Additional aspects e.g. based on histopathology may be of added value. CT values of Individual faecal samples using SYBR IS900 qPCR To look at the option to do a MAP specific qPCR on faecal material, targeting the IS900 fragment, our CT values ranged between 14 and 28. It is possible to do a MAP specific qPCR. Is there a correlation between qPCR and Serology (Paralisa)? 53 faecal and matched blood samples were compared. After classification of the results, as shown in Table 4, several similarities are apparent between qPCR CT category and Paralisa categories. A significant relationship between matched Cervine faecal (CT values) and Blood (Paralisa Johnin) values is verified with P <0.001. Taking this into account it may be interesting to perform both a qPCR and a “gold standard” radiometric culture assay (47) on the same sample and see how their results might relate to each other based on histological classification. Culture is considered as golden standard, but is has several disadvantages. A culturing system can be influenced by contamination or overgrowth of other gut flora. It can 27 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer also reduce the number of viable MAP by decontamination procedures (14, 33). MAP has a state of dormancy and is not always cultured even though the organisms are alive (47). These disadvantages may result in false negative results. qPCR assays on the opposite site, do not distinguish live MAP from dead MAP and there is a risk of lab contamination. CT values acquired by fecal samples compared to lymph node samples A comparison of 22 matched faecal and lymph node samples, showed that faecal qPCR results have a bigger range in CT value, and thus are a highly discriminatory value in diagnostics. The results also show that CT values acquired from lymph nodes are lower, indicating there is less mycobacterial DNA in the lymph node than there is in the faeces included in a similar amount of genomic DNA. The difference in CT values may also be attributed to the fact that only one lymph node was sampled, which might not be representative for the whole mesenteric tract. A faecal sample is a sample of everything that has been passing through the gut; therefore it may be more representative, although this is limited as it is single observation in time. Efficiency of the SYBR qPCR. Green compared to TaqMan assays. To improve the specificity and reliability of our results, TaqMan chemistry was used as with the SYBR Green chemistry some duplex-peak dissociation curves were obtained, due to the size and complexity of the population of DNA molecules recovered from a faecal DNA extraction. SYBR green based fluorescence assays can often yielded results which were difficult to interpret. This arises mainly because SYBR green dye binds to all double stranded DNA molecules in the sample and throughout the amplification reaction. TaqMan, as described in the introduction, only generates a signal from amplified product and not from nonspecific binding of sample DNA. No dissociation curve is required. The ability of using different colors to be detected on the probes allowed an internal amplification control(IAC) to be included to the assay. An IAC provides information if the assay on itself worked and if the DNA used was intact, as the target sequence of the IAC used in this study, GAPDH, is present in the cervine genome. For diagnostic quality assurance, the use of an IAC is thought to be mandatory in diagnostic PCR (17). For example, the study of Kawaji et al. (19) and Fang (12) do not have an IAC! By including an IAC control, false-negative results can be excluded. When there is no IAC there is no certainty if the qPCR on itself worked. Nineteen samples 28 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer (11 faecal and 9 lymph node samples) were tested with both SYBR Green and TaqMan chemistry 19 samples TaqMan gave higher PCR efficiency 1.9 / 1.8 SYBR and a 2.94 average lower CT value as we expected. What is the difference between F57 and IS900 in CT value? In this report the target sequences for the MAP specific qPCR are IS900 and F57. There is a study of Semret et al. (40) which argues that amplification of IS900-like sequences is not sufficient as a proxy for MAP. That Semret et al. determined the IS900 sequence conservation in a panel of 23 diverse isolates confirmed to be MAP, by the use of IS900 independent markers. The PCR products were sequenced. 100% identity to the IS900 elements of the published genome sequence of MAP strain K10 was acquired by all the 13 isolates of cattle used. 10 isolates of sheep showed that they had undergone polymorphisms at two loci. They observed that, because the origin of the samples was diverse, MAP isolates had highly uniform IS900 sequences and that polymorphisms described in other IS900 sequences (they found eight variants of the IS900 K10 strain previously described, differing by one or more single-nucleotide polymorphisms from the K10 strain (21) in public databases were not found in their diverse isolates. They state that because the fact that many studies relied solely on PCR detection of IS900 to identify MAP, and because of their found high sequence conservation in the MAP isolates, it is difficult to say whether these polymorphisms reflect true variants, or result instead of from technical sequencing errors. The study of Kawaji et al. (19) that detected C (316F) and S (Telford) strains of MAP by qPCR assay, reported no cross reactions with 51 other species of mycobacteria including 10 which contained IS900 like sequences. This suggests that it is possible to solely rely on the IS900 insertion element, when primers are specifically designed for MAP IS900 and to avoid IS900 -like sequences. In this study is not checked if the primers used give any cross reactions with other non-MAP mycobacterial strains. To confirm there are no cross reactions with IS900 like sequences, amplifications of non-MAP mycobacterial strains, containing IS900 sequences should be performed with the primers used in this study. Sequencing amplicons of the reaction should provide certainty about the product acquired. The study of Kawaji et al. (19) also reports that qPCR based on alternative genes, with lower copy numbers of them in the MAP genome, such as ISMav2, ISMap02 or F57, had lower sensitivities or required additional steps to obtain similar sensitivity to culture. In this study we used both, IS900 and F57 to might 29 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer improve PCR identification of MAP. Animals, tested IS900 positive, also tested F57 positive. The CT values acquired were in average 5.27 CT’s lower than the IS900 value found. This confirms that F57 has a lower sensitivity. Effect of laboratory contamination Results indicate that small amounts of qPCR-amplified product can influence the diagnostic outcome of the test. A contamination of 2 pL made a “low shedder” animal a “high shedder” animal. These 2 pL, based of figure 5, contain 75.000 genome copies of 5 fg. It is therefore imperative to use a closed amplification system and have a system to monitor template contamination. Uracil N-Glycosylase (UNG) can be used as an enzymatic decontaminant in PCR reactions. The use of UNG controls carry-over contamination and prevents false positive results from a previous qPCR run. dTTP is replaced by dUTP in the PCR-mix (Taq DNA-polymperase has the same affinity for dUTP as for dTTP) and pre-treatment of next PCR assays with UNG is performed. The UNG degrades specifically the dU-containing DNA, these chains cannot be used as template in a new amplification reaction. Pre-treatment with UNG in this procedure, prevents contamination from a previous PCR reaction where dUTP is used. Negative DNA is not degraded. UNG is degraded before the qPCR amplification begins by the high melting temperature of the first step in the PCR assay (22, 38). 30 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 6. Conclusion Development of a qPCR for Cervine faecal and tissue samples. qPCR obtained with SYBR green assay give CT values between 14 and 38. Endpoint PCR analysis showed amplicons of the expected fragment length, which confirms presence of IS900. A qPCR with the IS900 target sequence can be performed. To test analytical sensitivity CT values acquired (SYBR IS900) ranged from 17 to > 35 (negative cut-point). This range provides a base to classify results. The lowest quantity of MAP DNA which could be reliably detected was approximately 10 genome equivalents with a CT value 34 > 35.Analysis of Cervine matched faecal(qPCR) and blood samples(Paralisa Johnin) shows there is a significant correlation between these test results with a P < 0.001.Comparing matched lymph node samples to matched faecal samples shows that faecal CT values give a bigger range in CT values. Samples performed by TaqMan Chemistry were 2.94 CT lower compared to the SYBR green assay designed for the same target. PCR efficiency of TaqMan was superior to SYBR green. TaqMan 1.91 vs. 1.84 for SYBR green. TaqMan Chemistry also gave clearer amplification plots. Assays to compare F57 and IS900 CT values , IS900 was shown to have a consistently lower CT value for IS900. F57 is found to be a less sensitive target in this study. Laboratory contamination, investigated in this study concludes that a contamination of only 2pL makes a low shedder animal a high shedder animal, has a big effect on CT results. Continuation and future prospects According to the findings in this study, it is presumptive to continue the development of a proper working qPCR for Cervine faecal material. As TaqMan chemistry performes better than SYBR green assays, and the ability to use an IAC in TaqMan, TaqMan should be preferentially used. IS900 will give a more sensitive result than F57, although this may cost specificity. The specificity of the IS900 primers used should therefore be investigated as described in the discussion. With regard to the effects of laboratory contamination, it would be highly recommended to use a system that controls laboratory contamination. The study of Kawaji et al. (19) showed that quantitation of MAP DNA in faeces by the qPCR assay provides immediate information to estimate the risk of transmission from infected animals. They analysed DNA quantities detected by the qPCR, compared to histological classification, faecal samples representing sheep with multibacillary lesions showed significantly higher levels of MAP DNA than samples from sheep with paucibacillary lesions or no lesions. Diagnostically this may give the opportunity to help in JD control programmes. In this study we found a significant correlation between faecal qPCR results and blood samples tested by a validated Paralisa. Both reasons as mentioned above are a good reason to perform histological classification, as well as culture to matched qPCR samples in any following study, as it might will result in a statistical correlation with a golden standard test, in the future this may be a good option to provide complementary diagnostics. 31 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer word of thanks First of all I want to say “thank you” to everybody who Works at DRL of the Immunology & Microbiology Department of Otago University, for the support with my research and to make me feel home during my stay. Daily teatimes were a source of information and yummy refreshments. Thanks to Frank, who provided the possibility to do my research project in Dunedin, and for the nice tramp. Especially thanks to Rory who introduced me to the materials & methods DRL works with, explained me background in Molecular Biology, gave direction to my research, ordered stuff, read and corrected my report over and over again. Thanks to Simon for convincing deer farmers to participate in the research. Thanks to Alan for providing answers to my questions and for ordering lab stuff. Thanks to PhD students Sonia, Mark and Brooke for helping answering my questions and showing some of the Kiwi student life. Last, but not least, Ad, my supervisor from the Netherlands, for giving direction to my research and reading and correcting my report over and over again. Thanks to my family who assisted on the background. 32 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer References 1. Ausubel, F. M. 1990. Current protocols in molecular biology. Greene Publishing Associates and Wiley-Interscience, New York. 2. Bannantine, J. P., and J. R. Stabel. 2000. HspX is present within Mycobacterium paratuberculosis-infected macrophages and is recognized by sera from some infected cattle. Vet Microbiol 76:343-358. 3. Bustin, S. A. 2004. A-Z of quantitative PCR. International University Line, La Jolla, CA. 4. Bustin, S. A., V. Benes, T. Nolan, and M. W. Pfaffl. 2005. Quantitative real-time RT-PCR--a perspective. J Mol Endocrinol 34:597-601. 5. Chacon, O., L. E. Bermudez, and R. G. Barletta. 2004. Johne's disease, inflammatory bowel disease, and Mycobacterium paratuberculosis. Annu Rev Microbiol 58:329-363. 6. Collins, D. M., D. M. Gabric, and G. W. De Lisle. 1989. Identification of a repetitive DNA sequence specific to Mycobacterium paratuberculosis. FEMS Microbiol Lett 51:175-178. 7. Collins, M. T. 1996. Diagnosis of paratuberculosis. Vet Clin North Am Food Anim Pract 12:357-371. 8. Cook, K. L., and J. S. Britt. 2007. Optimization of methods for detecting Mycobacterium avium subsp. paratuberculosis in environmental samples using quantitative, real-time PCR. J Microbiol Methods 69:154-160. 9. Cousins, D. V., R. Whittington, I. Marsh, A. Masters, R. J. Evans, and P. Kluver. 1999. Mycobacteria distenct from Mycobacterium avium subsp. paratuberculosis isolated from the faeces of ruminants possess IS900-like sequences detectable IS900 polymerase chain reaction: implications for diagnosis. Mol Cell Probes 13:431-442. 10. de Lisle, G. W., G. F. Yates, and H. Montgomery. 2003. The emergence of Mycobacterium paratuberculosis in farmed deer in New Zealand - a review of 619 cases. N Z Vet J 51:58-62. 11. Englund, S., G. Bolske, and K. E. Johansson. 2002. An IS900-like sequence found in a Mycobacterium sp. other than Mycobacterium avium subsp. paratuberculosis. FEMS Microbiol Lett 209:267-271. 12. Fang, Y., W. H. Wu, J. L. Pepper, J. L. Larsen, S. A. Marras, E. A. Nelson, W. B. Epperson, and J. Christopher-Hennings. 2002. Comparison of real-time, quantitative PCR with molecular beacons to nested PCR and culture methods for detection of Mycobacterium avium subsp. paratuberculosis in bovine fecal samples. J Clin Microbiol 40:287-291. 13. Green, E. P., M. L. Tizard, M. T. Moss, J. Thompson, D. J. Winterbourne, J. J. McFadden, and J. Hermon-Taylor. 1989. Sequence and characteristics of IS900, an 33 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer insertion element identified in a human Crohn's disease isolate of Mycobacterium paratuberculosis. Nucleic Acids Res 17:9063-9073. 14. Griffin, J. F., E. Spittle, C. R. Rodgers, S. Liggett, M. Cooper, D. Bakker, and J. P. Bannantine. 2005. Immunoglobulin G1 enzyme-linked immunosorbent assay for diagnosis of Johne's Disease in red deer (Cervus elaphus). Clin Diagn Lab Immunol 12:1401-1409. 15. Gumbrell, R. C. 1986. Johne's Disease in Deer. Surveillance 13:15-16. 16. Harris, N. B., and R. G. Barletta. 2001. Mycobacterium avium subsp. paratuberculosis in Veterinary Medicine. Clin Microbiol Rev 14:489-512. 17. Hoorfar, J., N. Cook, B. Malorny, M. Wagner, D. De Medici, A. Abdulmawjood, and P. Fach. 2003. Making internal amplification control mandatory for diagnostic PCR. J Clin Microbiol 41:5835. 18. Irenge, L. M., K. Walravens, M. Govaerts, J. Godfroid, V. Rosseels, K. Huygen, and J. L. Gala. 2009. Development and validation of a triplex real-time PCR for rapid detection and specific identification of M. avium sub sp. paratuberculosis in faecal samples. Vet Microbiol 136:166-172. 19. Kawaji, S., D. L. Taylor, Y. Mori, and R. J. Whittington. 2007. Detection of Mycobacterium avium subsp. paratuberculosis in ovine faeces by direct quantitative PCR has similar or greater sensitivity compared to radiometric culture. Vet Microbiol 125:36-48. 20. Kennedy, D. J., and G. Benedictus. 2001. Control of Mycobacterium avium subsp. paratuberculosis infection in agricultural species. Rev Sci Tech 20:151-179. 21. Li, L., J. P. Bannantine, Q. Zhang, A. Amonsin, B. J. May, D. Alt, N. Banerji, S. Kanjilal, and V. Kapur. 2005. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci U S A 102:12344-12349. 22. Longo, M. C., M. S. Berninger, and J. L. Hartley. 1990. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene 93:125-128. 23. Machackova-Kopecna, M., M. Bartos, M. Straka, V. Ludvik, P. Svastova, J. Alvarez, J. Lamka, I. Trcka, F. Treml, I. Parmova, and I. Pavlik. 2005. Paratuberculosis and avian tuberculosis infections in one red deer farm studied by IS900 and IS901 RFLP analysis. Vet Microbiol 105:261-268. 24. Mackintosh, C. G. 2002. Summary of a Johne’s disease session: NADVet Conference, Austin, Texas, 19-20 February 2002. Proceedings of a Deer Course for Veterinarians. Deer Branch of the New Zealand Veterinary Association 19:19-28. 25. Mackintosh, C. G., and G. W. De Lisle. 1998. Johne’s Disease: The current situation in New Zealand Deer. Proceedings of a Deer Course for Veterinarians. Deer Branch of the New Zealand Veterinary Association 15:175-182. 34 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 26. Mackintosh, C. G., G. W. de Lisle, D. M. Collins, and J. F. Griffin. 2004. Mycobacterial diseases of deer. N Z Vet J 52:163-174. 27. Mackintosh, C. G., T. Qureshi, K. Waldrup, R. E. Labes, K. G. Dodds, and J. F. Griffin. 2000. Genetic resistance to experimental infection with Mycobacterium bovis in red deer (Cervus elaphus). Infect Immun 68:1620-1625. 28. McKenna, S. L., G. P. Keefe, A. Tiwari, J. VanLeeuwen, and H. W. Barkema. 2006. Johne's disease in Canada part II: disease impacts, risk factors, and control programs for dairy producers. Can Vet J 47:1089-1099. 29. Millar, D., J. Ford, J. Sanderson, S. Withey, M. Tizard, T. Doran, and J. Hermon-Taylor. 1996. IS900 PCR to detect Mycobacterium paratuberculosis in retail supplies of whole pasteurized cows' milk in England and Wales. Appl Environ Microbiol 62:3446-3452. 30. Mobius, P., H. Hotzel, A. Rassbach, and H. Kohler. 2008. Comparison of 13 singleround and nested PCR assays targeting IS900, ISMav2, f57 and locus 255 for detection of Mycobacterium avium subsp. paratuberculosis. Vet Microbiol 126:324333. 31. Paustian, M. L., A. Amonsin, V. Kapur, and J. P. Bannantine. 2004. Characterization of novel coding sequences specific to Mycobacterium avium subsp. paratuberculosis: implications for diagnosis of Johne's Disease. J Clin Microbiol 42:2675-2681. 32. Poupart, P., M. Coene, H. Van Heuverswyn, and C. Cocito. 1993. Preparation of a specific RNA probe for detection of Mycobacterium paratuberculosis and diagnosis of Johne's disease. J Clin Microbiol 31:1601-1605. 33. Reddacliff, L. A., A. Vadali, and R. J. Whittington. 2003. The effect of decontamination protocols on the numbers of sheep strain Mycobacterium avium subsp. paratuberculosis isolated from tissues and faeces. Vet Microbiol 95:271-282. 34. Rodriguez-Lazaro, D., M. D'Agostino, A. Herrewegh, M. Pla, N. Cook, and J. Ikonomopoulos. 2005. Real-time PCR-based methods for detection of Mycobacterium avium subsp. paratuberculosis in water and milk. Int J Food Microbiol 101:93-104. 35. Sambrook, J., D. W. Russell, N. Irwin, K. A. Janssen, and T. Maniatis. 2001. Molecular cloning : a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. 36. Sanderson, J. D., M. T. Moss, M. L. V. Tizard, and J. Hermontaylor. 1991. Pcr Detection of Mycobacterium-Paratuberculosis in Crohns-Disease Tissue DNA Extracts. Proceedings of the Third International Colloquium on Paratuberculosis:201207. 37. Sartor, R. B. 2005. Does Mycobacterium avium subspecies paratuberculosis cause Crohn's disease? Gut 54:896-898. 35 Development of a qPCR assay for Mycobacterium avium subsp. Paratuberculosis (MAP) in Deer 38. Schmidt, M., B. Frey, K. Kaluza, and H. Sobek. 1996. Application of Heat-Labile Uracil-DNA Glycosylase in Improved Carryover Prevention Technique. Biochemica 2:13-15. 39. Schonenbrucher, H., A. Abdulmawjood, K. Failing, and M. Bulte. 2008. New triplex real-time PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in bovine feces. Appl Environ Microbiol 74:2751-2758. 40. Semret, M., C. Y. Turenne, and M. A. Behr. 2006. Insertion sequence IS900 revisited. J Clin Microbiol 44:1081-1083. 41. Stabel, J. R., and J. P. Bannantine. 2005. Development of a nested PCR method targeting a unique multicopy element, ISMap02, for detection of Mycobacterium avium subsp. paratuberculosis in fecal samples. J Clin Microbiol 43:4744-4750. 42. Strommenger, B., K. Stevenson, and G. F. Gerlach. 2001. Isolation and diagnostic potential of ISMav2, a novel insertion sequence-like element from Mycobacterium avium subspecies paratuberculosis. FEMS Microbiol Lett 196:31-37. 43. Taddei, S., C. Robbi, C. Cesena, I. Rossi, E. Schiano, N. Arrigoni, G. Vicenzoni, and S. Cavirani. 2004. Detection of Mycobacterium avium subsp. paratuberculosis in bovine fecal samples: comparison of three polymerase chain reaction-based diagnostic tests with a conventional culture method. J Vet Diagn Invest 16:503-508. 44. Thibault, V. C., M. Grayon, M. L. Boschiroli, C. Hubbans, P. Overduin, K. Stevenson, M. C. Gutierrez, P. Supply, and F. Biet. 2007. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol 45:2404-2410. 45. Tiwari, A., J. A. VanLeeuwen, S. L. McKenna, G. P. Keefe, and H. W. Barkema. 2006. Johne's disease in Canada Part I: clinical symptoms, pathophysiology, diagnosis, and prevalence in dairy herds. Can Vet J 47:874-882. 46. Whipple, D. L., P. A. Kapke, and P. R. Andersen. 1992. Comparison of a commercial DNA probe test and three cultivation procedures for detection of Mycobacterium paratuberculosis in bovine feces. J Vet Diagn Invest 4:23-27. 47. Whittington, R. J., I. Marsh, S. McAllister, M. J. Turner, D. J. Marshall, and C. A. Fraser. 1999. Evaluation of modified BACTEC 12B radiometric medium and solid media for culture of Mycobacterium avium subsp. paratuberculosis from sheep. J Clin Microbiol 37:1077-1083. 48. Whittington, R. J., D. J. Marshall, P. J. Nicholls, I. B. Marsh, and L. A. Reddacliff. 2004. Survival and dormancy of Mycobacterium avium subsp. paratuberculosis in the environment. Appl Environ Microbiol 70:2989-3004. 36