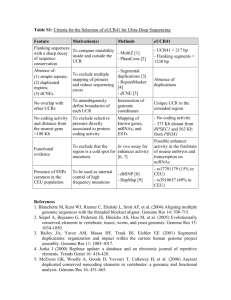
Supplementary figure legends - Proceedings of the Royal Society B
advertisement

Supplementary Figure 1. Derived duplications and deletions versus duplications and deletions relative to a reference genome. a) A duplication detected relative to a reference genome may in fact be a deletion allele of previously duplicated sequence, with the reference genome having the deletion allele. Note that for this to be the case, the duplication must have been recent enough for DNA from both copies present in a sample lacking the deletion allele to exhibit hybridization to the set of probes corresponding to the single copy present in the reference genome. This implies that apparent duplications that are actually deletions of duplicated sequences will rarely be detected unless certain regions of the genome have high enough mutations rates for recent duplicates to have an appreciable chance of being deleted. b) A deletion of previously duplicated sequence detected relative to a reference genome may in fact be a duplication present in the reference genome but not in all samples. Note that these will only be detected when the duplicates are dissimilar enough to be probed separately. Thus, cases like the bottom of panel b) will likely rarely be detected. Supplementary Figure 2. Allele frequency spectrum of CNVs detected in 15 inbred Drosophila melanogaster lines by Emerson et al. (2008). The data are shown before and after correcting for the ascertainment bias due to undetected mutations present in the reference genome. The correction in this case is to divide each bin by 1-p. When the reference genome is an outbred individual, corrections are more complicated. If the “presence” allele is always represented when individuals are heterozygous for duplications or deletions, we could assume that the only missed sequences would be homozygous deletions in the reference individual or duplications present in either the homozygous or heterozygous state in the reference individual. This means that the correction would simply be to divide by 1-p2 in the case deletions and to divide by 1-(1p)2 in the case of duplications. However, given the almost unfathomable proclivities of assembly software, we may never be able to confidently say that one allele or another makes it into the reference consistently. In addition, in cases where multiple outbred individuals were sequenced in order to construct the reference genome—as in humans— there is no straightforward correction possible.